
記住我
Among the multitude of molecularly, morphologically and functionally diverse cells in the human brain is a quite prominent but numerically very small population, comprising roughly 1% or in average 500,000 of the brain’s nerve cells: the dopamine (DA)-synthesizing neurons in the human ventral midbrain (VM)/brainstem (Pakkenberg et al., 1991). DA is a catecholamine synthesized as a direct derivative of the amino acid tyrosine by the rate-limiting enzyme tyrosine hydroxylase (TH) and the dopa decarboxylase (DDC, or aromatic L-amino acid decarboxylase/AADC) in a common biosynthetic pathway with other catecholamines, such as noradrenaline and adrenaline (Meiser et al., 2013). Because of its binding to a family of at least five different metabotropic DA receptors (DRD1-5) coupled to either stimulatory (Gs) or inhibitory (Gi) G-proteins, DA exerts a neuromodulatory function in the brain (Money and Stanwood, 2013; Zhai et al., 2019). In this review, the focus is set exclusively on the midbrain dopaminergic system due to its prominent role in animal and human behavior.
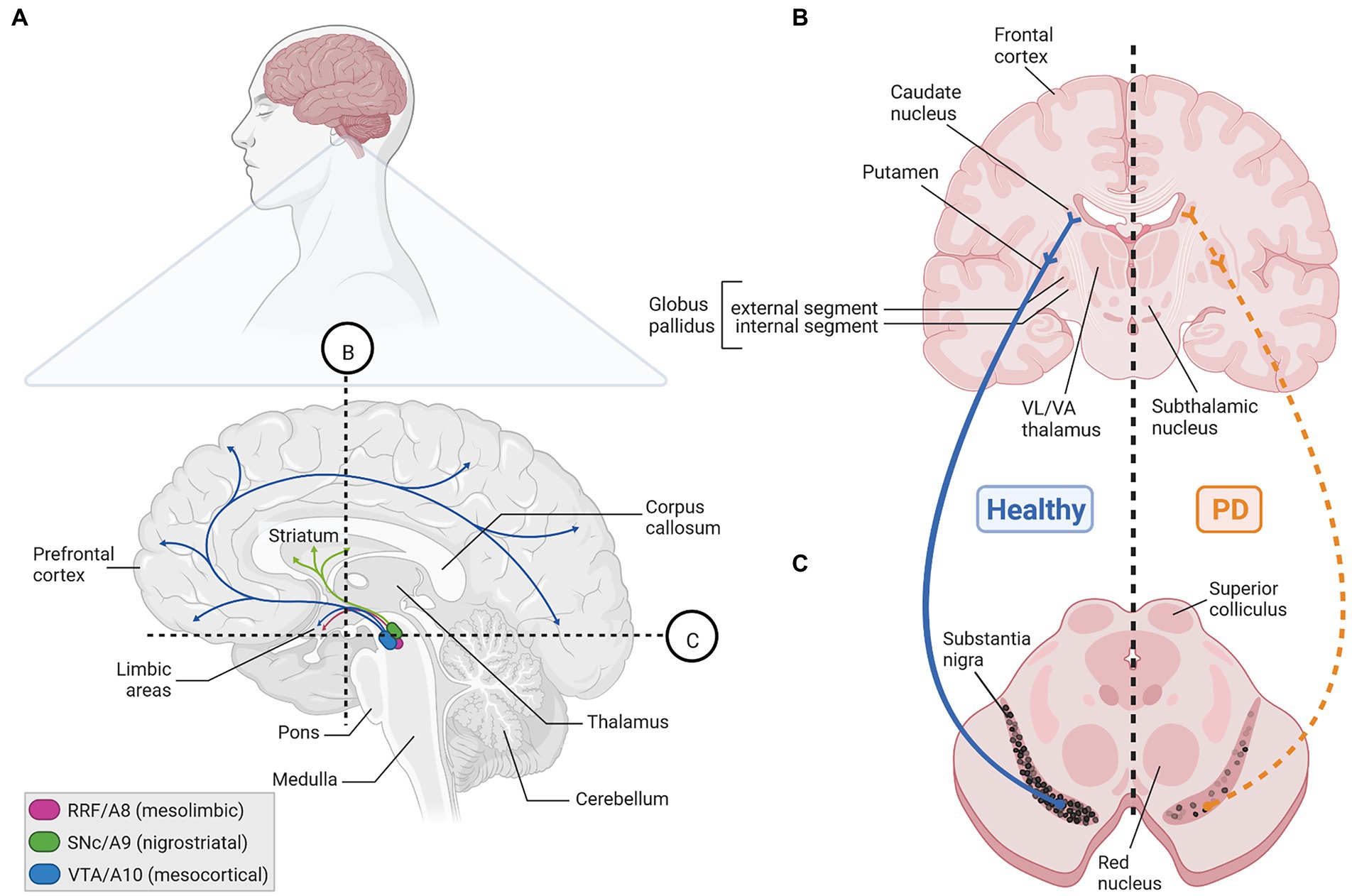
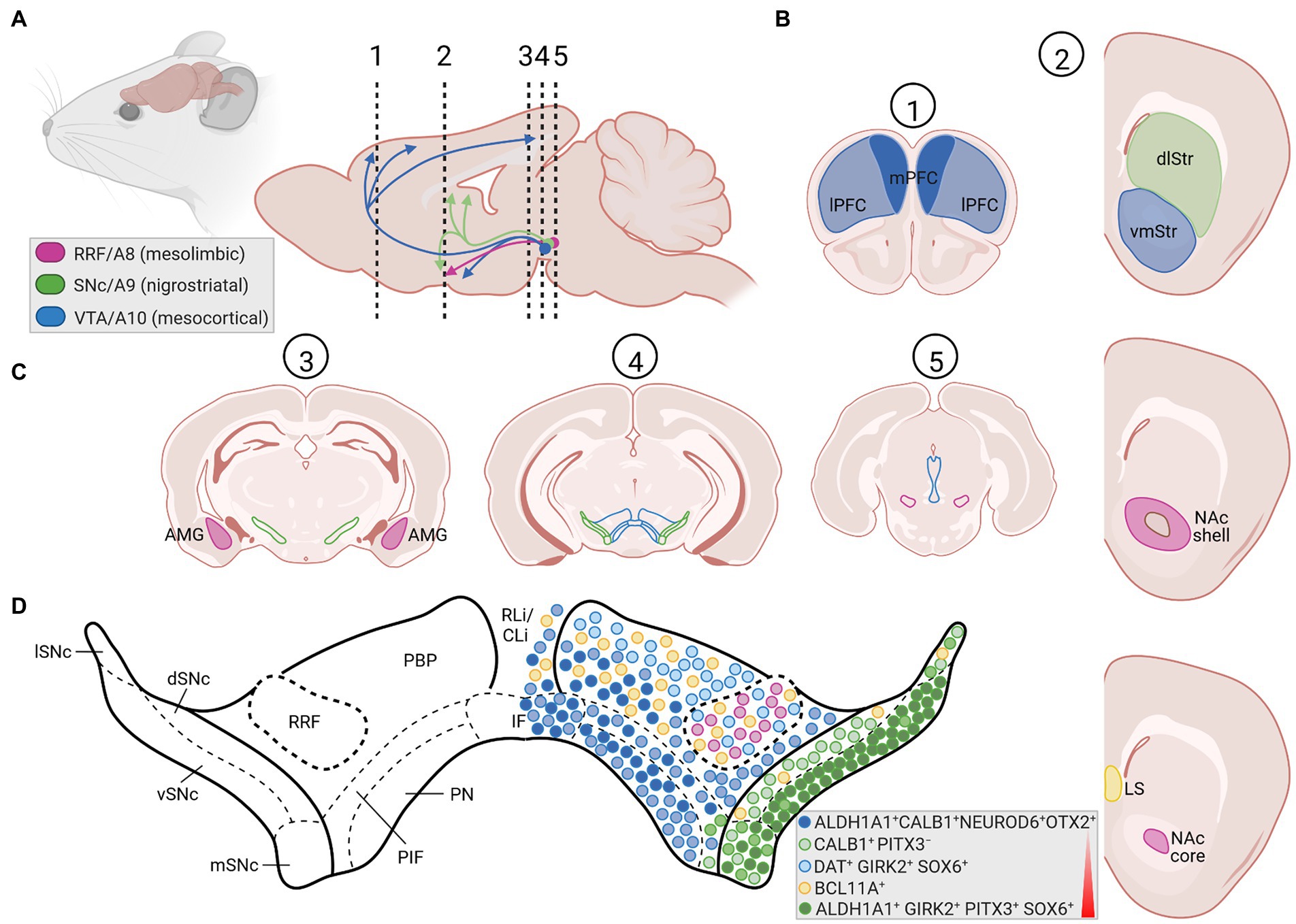
Three major cluster of DA-synthesizing neurons are typically found in the mammalian VM, collectively named midbrain dopaminergic (mDA) neurons. These comprise, in a caudal to rostral numbering, the mDA neurons located in the retrorubral field (RRF, A8 group), substantia nigra pars compacta (SNc, A9 group) and ventral tegmental area (VTA, A10 group; Bjorklund and Dunnett, 2007; Figures 1A, 2A,C). In the rodent brain, the SNc DA neurons or A9 group consist of the dorsal, ventral, medial and lateral tier, whereas the VTA DA neurons or A10 group are made up by the parabrachial pigmented nucleus (PBP), paranigral nucleus (PN), interfascicular nucleus (IF), parainterfascicular nucleus (PIF), and rostral linear nucleus (RLi) and caudal linear nucleus (CLi) of the raphe (Morales and Margolis, 2017; Figures 2C,D). Although this classification is still widely used in the field, extensive analyses of the rodent and human mDA systems revealed that each of these larger mDA groups are made up by molecularly, morphologically and functionally diverse mDA neurons that can be further subdivided into several subgroups, in particular in the SNc and VTA (Roeper, 2013; Poulin et al., 2020). The cell bodies of the SNc DA neurons send their axons primarily to the dorsolateral striatum in rodents (caudate-putamen in primates) within the so-called nigrostriatal pathway, which is a part of the basal ganglia circuitry encompassing also the globus pallidus (internal and external segment) and subthalamic nucleus (Figures 1B,C, 2A,B). The release of DA from these neurons modulates the activity of their target cells in the striatum for the control of voluntary movements and motor learning (Arber and Costa, 2022). The VTA and RRF DA neurons innervate the prefrontal cortex (PFC; mesocortical pathway) and limbic regions of the brain (mesolimbic pathway), including the ventromedial striatum (nucleus accumbens, NAc), olfactory tubercle (OT), amygdala (AMG), septum, cingulate, and perirhinal cortex (Figures 1A, 2A,B). Accordingly, DA release from these neurons is implicated in the control and modulation of cognitive, emotive/affective, motivational/salient and rewarding behaviors (Cox and Witten, 2019; Arber and Costa, 2022). Because of these particular functions, the mDA neurons have gained particular attention in the clinical context: the age-dependent and progressive degeneration of the neuromelanin-containing (dark pigmented) vSNc DA neurons in the human brain is considered a hallmark of PD (Figure 1C), whereas the dysregulation of DA release from the VTA DA neurons is implicated in the pathogenesis of severe neuropsychiatric human disorders, including schizophrenia and substance use disorders (SUD). In fact, insights into the functions of the mDA system in the human context have come primarily from the study and treatment of these diseases. The focus of this review is the particular vulnerability of the mDA neurons to degeneration, and thus restricted to their closer examination in the context of PD.
Figure 1. The human mDA system in the adult healthy and PD brain. (A) Sagittal view (upper) and enlarged parasagittal section (lower) of the adult human brain, showing the location of the cell bodies (ovals) from the A8 (RRF, pink), A9 (SNc, green), and A10 (VTA, blue) mDA clusters in the brainstem, and their projections (arrows) to the limbic areas (mesolimbic pathway), striatum (nigrostriatal pathway) and PFC (mesocortical pathway). Note that both the VTA and RRF DA neurons project to limbic areas in the mesolimbic pathway. Labeling of other structures (hypothalamus, etc.) and limbic regions, such as the NAc, olfactory tubercle, amygdala, septum, cingulate and perirhinal cortex, in this sagittal view have been omitted for clarity. (B,C) Cross-sections at the levels of the dotted lines in (A) through the human forebrain (B) and midbrain/brainstem (C) depicting the location of the projection areas and cell bodies, respectively, of the human SNc DA subset. In the healthy human brain (left side), dark pigmented (neuromelanin-containing) SNc DA neurons extend their axons into the caudate putamen, where they synapse onto their immediate target cells, the striatal GABAergic medium spiny neurons (MSN) and cholinergic and GABAergic fast spiking (FS) interneurons (INs). The MSN neurons project to the internal or external segment of the globus pallidus which, in turn, are connected with the ventrolateral (VL) and ventroanterior (VA) thalamus or subthalamic nucleus in what is known as the direct or indirect pathway, respectively, of the basal ganglia (not depicted here). Retrograde degeneration of the axons and subsequently of the pigmented cell bodies in the SNc (right side) is one key histopathological feature of the PD brain. Created with BioRender.com.
Figure 2. The rodent mDA system in the adult brain. (A) Sagittal view (left) and enlarged midsagittal section (right) of the adult rodent (mouse, rat) brain, showing the location of the cell bodies (circles) from the A8 (RRF, pink), A9 (SNc, green), and A10 (VTA, blue) mDA clusters in the VM, and their projections (arrows) to the limbic areas (mesolimbic pathway), striatum (nigrostriatal pathway) and PFC (mesocortical pathway). Note that both the VTA and RRF DA neurons project to limbic areas in the mesolimbic pathway. The dotted and numbered lines indicate the position of the cross sections shown in (B,C). (B) Cross-sections of the rodent forebrain at the level of the dotted lines #1 and 2 in (A), depicting the location of the medial (mPFC, dark blue area) and lateral (lPFC, light blue area) PFC (#1) as one target area of the VTA DA subset, of the dorsolateral striatum (dlStr, green area), ventromedial striatum (vmStr, blue area), NAc shell or NAc core (pink area; right panel #2) as preferential target areas of the SNc, VTA and RRF DA subsets, respectively, and of the lateral septum (LS, yellow area) innervated by NEUROD6-and BCL11A-expressing mDA neurons. (C) Cross-sections at different rostro-caudal levels of the rodent midbrain, indicated by the dotted lines #3, 4 and 5 in (A), indicating the positions of the SNc (green lines), VTA (blue lines) and RRF (pink lines) DA subpopulations, as well as the location of the amygdala (AMG, pink area) as another target area in the mesolimbic pathway. Labeling of other brain regions in these sagittal and coronal views have been omitted for clarity. (D) Enlargement of the mDA region in the medial rodent VM [level of the dotted line and cross-section #4 in (A,C), respectively], showing the SNc (ventral tier, vSNc; dorsal tier, dSNc; medial tier, mSNc; and lateral tier, lSNc), VTA (PBP, parabrachial pigmented nucleus; PN, paranigral nucleus; IF, interfascicular nucleus; PIF, parainterfascicular nucleus; RLi, rostral linear nucleus; CLi, caudal linear nucleus of the raphe), and RRF subdivisions. Colored circles depict the approximate positions and proportions of mDA neurons with distinctive gene expression signatures (legend in bottom right corner) within these subdivisions, as summarized in this article. Colors and shadings indicate the increasing vulnerability (red triangle) of these mDA subsets to neurodegeneration. Intermediate shadings denote mDA subsets not defined here. Created with BioRender.com.
PD is the second most common age-dependent neurodegenerative disorder after Alzheimer’s Disease currently affecting an estimated more than 6 million people worldwide, with an increasing prevalence of 1 to 3% above the age of 60 and 80, respectively (Tysnes and Storstein, 2017; Feigin et al., 2019). The increased life expectancy, particularly in the high and middle income countries, let some authors to call out actions against an expected “PD pandemic” already in 2018 (Dorsey and Bloem, 2018). Due to the ongoing global COVID-19 pandemic, which can also affect directly or indirectly the developing and adult human brain (Iadecola et al., 2020; Shook et al., 2022), it is at present unclear how the actual numbers of PD cases will develop in the future. PD is characterized by three cardinal symptoms: bradykinesia (slowness of movement), rigidity and resting tremor (Tysnes and Storstein, 2017; Balestrino and Schapira, 2020; Blesa et al., 2022). Bradykinesia can progress to hypokinesia/akinesia (partial or complete loss of movement), and other motor and non-motor/prodromal symptoms, such as postural instability, hyposmia, constipation and sleep disorder, typically accompany the disease, thus causing a broad disability in PD patients (Balestrino and Schapira, 2020; Blesa et al., 2022). The motor symptoms of PD usually arise and develop asymmetrically in the patients, and the initial mDA neuropathology spreads to other neurons and regions of the human brain during the course of the disease (Blesa et al., 2022). The cardinal PD symptoms are caused mainly by the lack of DA in the striatum (Figures 1B,C), leading (in a very simplistic view) to the hyperactivation of the indirect basal ganglia pathway that inhibits voluntary motor routines (Charvin et al., 2018; Blesa et al., 2022). The typical PD medication thus aims at restoring the DA supply in the striatum, primarily by the systemic administration of the DA precursor L-3,4-dihydroxyphenylalanine (L-DOPA, synthesized by TH from L-tyrosine). In contrast to DA, L-DOPA crosses the blood–brain-barrier (BBB) and reaches the remaining SNc DA axon terminals in the striatum, where it is metabolized into DA (Connolly and Lang, 2014; Balestrino and Schapira, 2020). Alternative but less frequently employed symptomatic treatments of PD, which also aim at reestablishing the normal activity of the basal ganglia circuitry, include deep brain stimulation and the not yet implemented (in the clinical routine) gene therapy or transplantation of mDA precursors into the diseased striatum (Blits and Petry, 2016; McIntyre and Anderson, 2016; Balestrino and Schapira, 2020; Harris et al., 2020; Parmar et al., 2020). Due to the unclear etiopathology of PD, particularly of the 90%–95% idiopathic (“sporadic,” because no cause is known) cases, disease-preventing, modifying, or halting treatments are still not available for this neurodegenerative disorder (Charvin et al., 2018; Balestrino and Schapira, 2020). The late diagnosis of PD (usually when at least 50% of the SNc DA neurons have already died; Surmeier et al., 2017; Blesa et al., 2022), puts forward an urgent need of early diagnostic biomarkers enabling a timely intervention in the expected future rise of PD cases (Balestrino and Schapira, 2020; Chmielarz and Saarma, 2020; Tönges et al., 2022).
The single major risk factor for PD is age (Collier et al., 2011; Tysnes and Storstein, 2017; Balestrino and Schapira, 2020). The mDA neurodegenerative process, particularly in the late-onset idiopathic cases, is thus thought to begin well beyond the juvenile and young adult age (Collier et al., 2011; Blesa et al., 2022). Several pathogenic mechanisms have been discussed, some of which appear to be shared by different human neurodegenerative diseases (Gan et al., 2018). Among these, the accumulation and defective proteasomal clearance of misfolded proteins, such as alpha-synuclein (SNCA), dysfunctional mitochondria leading to increased oxidative stress, reduced clearance of these and other defective organelles via the mitophagic/autophagic-lysosomal pathway (ALP), as well as a dysregulated calcium (Ca2+) homeostasis are considered as the main culprit, which may be exacerbated by a local or systemic immune response (Michel et al., 2016; Gan et al., 2018; Johnson et al., 2019; Panicker et al., 2021). These pathogenic impacts, either individually or altogether, ultimately lead to the apoptotic (programmed) cell death of the SNc DA neurons (Erekat, 2018). The precise reasons why SNc DA neurons are more affected in PD than VTA DA neurons, and why these mDA neurons appear to succumb earlier to degeneration compared to other neurons of the human brain, are not yet clear, but have been linked to increased metabolic energy requirements and a higher Ca2+ dependency of the SNc DA neurons [section 2 (Surmeier et al., 2017; Surmeier, 2018; Ni and Ernst, 2022) and references therein]. Moreover, these events are triggered by either a genetic predisposition (in around 5%–10% of PD cases) or environmental impacts (including the exposure to pesticides, heavy metals and infectious agents), or both (Johnson et al., 2019). The genetic (familial) forms of PD (fPD) are caused by mutations in PD-associated genes, so-called PARK loci (Hernandez et al., 2016; Benson and Huntley, 2019; Vázquez-Vélez and Zoghbi, 2021). Strikingly, PARK genes whose mutations lead to early-onset PD symptoms (i.e., motor symptoms appear during the first to third decade of life) are expressed in the mammalian embryo in a ubiquitous pattern that is not restricted to the VM or mDA system, whereas PARK genes causing mostly late-onset PD symptoms [i.e., motor symptoms appear from the fifth decade of life onward, thus resembling idiopathic PD (iPD)] when mutated, start to be expressed only slightly later in the target areas (striatum, cortex) of the mDA neurons (Benson and Huntley, 2019). The latter group include the PARK1/4 locus encoding mutant forms of SNCA and the PARK8 locus encoding leucine rich repeat kinase 2 (LRRK2). Abnormal accumulation and fibril formation of SNCA lead to the appearance of Lewy bodies and neurites in the PD brain as one of its histopathological hallmarks, whereas LRRK2 is a multifaceted protein with as yet ill-defined functions in the brain whose mutant variants are the second most common genetic risk factor for iPD (Benson and Huntley, 2019; Panicker et al., 2021; Vázquez-Vélez and Zoghbi, 2021). Mutant mice for several of these PARK genes display measurable alterations in neurogenesis and synaptic plasticity within the corticostriatal circuitry and other DA-modulated brain circuits at early postnatal or young adult ages, which in some cases also lead to detectable behavioral deficits (Le Grand et al., 2015; Benson and Huntley, 2019; Huntley and Benson, 2020). This indicates that mutations in at least some of the PARK genes impair the normal development of the mammalian brain, and strongly suggest that the pathogenic process in PD might begin much earlier than anticipated in the prevailing view. A defective mDA neuron development and maintenance resulting in functionally compromised or reduced numbers of SNc DA neurons could be another early (or the earliest) predisposing factor for the progression to PD in the aging individual or under additional adverse environmental and/or genetic conditions. These early mDA deficits are compensated over a significant amount of time by an enhanced DA metabolism or signaling in the remaining cells (Blesa et al., 2022). In the terminology of (Johnson et al., 2019), deficits in mDA and particularly SNc DA neuron development and maintenance might be considered an additional type of “early” facilitator for the emergence of PD, which per se is not sufficient to cause this disease.
This article summarizes mostly rodent and eventually primate in vivo studies providing strong evidences for the requirement and neuroprotective role of “developmental factors” in the adult mammalian VM and mDA system. Only those secreted signaling factors and nuclear transcription factors (TFs) active in the mDA domain during prenatal/embryonic development and additionally during adulthood are considered as “developmental factors.” The usability of these factors for preventive or therapeutic interventions in the PD brain is also reviewed, but the myriad of in vitro studies in this regard are not addressed here. The involvement of these signaling and/or transcriptional cascades in ongoing neurodegenerative processes associated with PD are not discussed as they were recently reviewed by others (Jha et al., 2022). This article does not delve into the mDA ontogenic and PD-associated programmed cell death (apoptosis; Burke, 2004; Savitt et al., 2005; Erekat, 2018; Robinson et al., 2018) or the role of “classical” neurotrophic factors, such as glial cell derived neurotrophic factor (GDNF), brain derived neurotrophic factor (BDNF), cerebral dopamine neurotrophic factor and mesencephalic astrocyte derived neurotrophic factor (Chmielarz and Saarma, 2020), in this context. The reader is referred to the many excellent reviews on these subjects, of which only the most recent ones are cited in this article.
2. Developmental mDA neuron deficits as another hit or facilitator in the “multiple hit” or “stochastic acceleration” hypotheses of Parkinson’s disease?The idea that a faulty generation, wiring and/or survival, leading to a reduced number of mDA and particularly SNc DA or other neurons already at the earliest stages of life (infancy and youth), might contribute to the emergence of PD and other neurodegenerative disorders in the aging individual is not new and has been proposed by several authors over the last 20 years [(Barzilai and Melamed, 2003; Carvey et al., 2006; Barlow et al., 2007; Ben-Ari, 2008; Le et al., 2009; Von Linstow et al., 2020) and references therein]. This hypothesis was mainly supported by the disentangling of the genetic basis of mDA neuron development in the mammalian embryo (section 3), and the discovery of typically single nucleotide polymorphisms increasing the risk for iPD and fPD that are associated with some of the most prominent genes in this context, such as EN1 (Fuchs et al., 2009; Haubenberger et al., 2011), EN2 (Rissling et al., 2009), NURR1 (NR4A2; Xu et al., 2002; Le et al., 2003; Wellenbrock et al., 2003; Zheng et al., 2003; Grimes et al., 2006; Jacobsen et al., 2008; Sleiman et al., 2009), PITX3 (Fuchs et al., 2009; Bergman et al., 2010; Guo et al., 2011; Haubenberger et al., 2011; Le et al., 2011), LMX1A/B (Bergman et al., 2009), and FGF20 (van der Walt et al., 2004; Mizuta et al., 2008; Wang et al., 2008; Pan et al., 2012; Nalls et al., 2019). Most of these point mutations are located in non-coding regions and are thus not expected to impact the function of the corresponding proteins, but rather—if at all—their expression levels (gene dosage; Von Linstow et al., 2020). This suggested that the developmental deficits that might be associated with an increased risk for PD must be subtle and/or masked by compensatory mechanisms during a relevant period of life (Blesa et al., 2022). More recent data indicated that the initiation of PARK gene expression in the embryonic rodent brain correlates with the beginning of mDA neurogenesis, and is in some cases strongest in the developing VM (mDA domain) or striatum (mDA target region) [(Sulzer, 2007; Le Grand et al., 2015; Benson and Huntley, 2019) and references therein]. They provided further support for an etiological component of PD that is rooted in early developmental deficits, at least in some cases (Von Linstow et al., 2020), although the precise functions of the corresponding PARK proteins in the healthy brain and especially at these early developmental stages are only beginning to be unraveled (Le Grand et al., 2015; Benson and Huntley, 2019). Nevertheless, PD does not fulfill the criteria of a typical neurodevelopmental disorder because its earliest diagnosis is restricted to very rare cases in a juvenile age (in their teens), and most cases, especially the vast majority of idiopathic cases, are diagnosed only at an advanced age (from their fifties onward; Tysnes and Storstein, 2017). Even if the diagnosis of PD is preceded by a considerable prodromal or pre-symptomatic period of up to 20 years (Blesa et al., 2022), the initiation of the pathogenic process would still be confined to the midlife age in the majority of PD cases (Collier et al., 2011; Von Linstow et al., 2020). A defective development and early survival of the mDA neurons might predispose to the later emergence of PD by acting as an additional risk factor or early “facilitator” for the disease (Figure 3). The term “facilitator” is used in a slightly deviant definition from (Johnson et al., 2019) as a factor that promotes but is not necessary and precedes additional “triggers” to initiate the disease process. This is because there is no human genetic or epidemiologic evidence so far for mutations in developmental genes or deficient developmental processes being a necessary prerequisite for the emergence of PD, and because developmental deficits inherently have to be one of the first if not the first insult in this context. Ultimately, “facilitators,” “triggers,” and “aggravators” have to converge in a probably self-reinforcing process described by the “multiple hit hypothesis” (Carvey et al., 2006; Sulzer, 2007) or, more recently, by the “stochastic acceleration hypothesis” (Collier et al., 2011) to cause the appearance of PD symptoms.
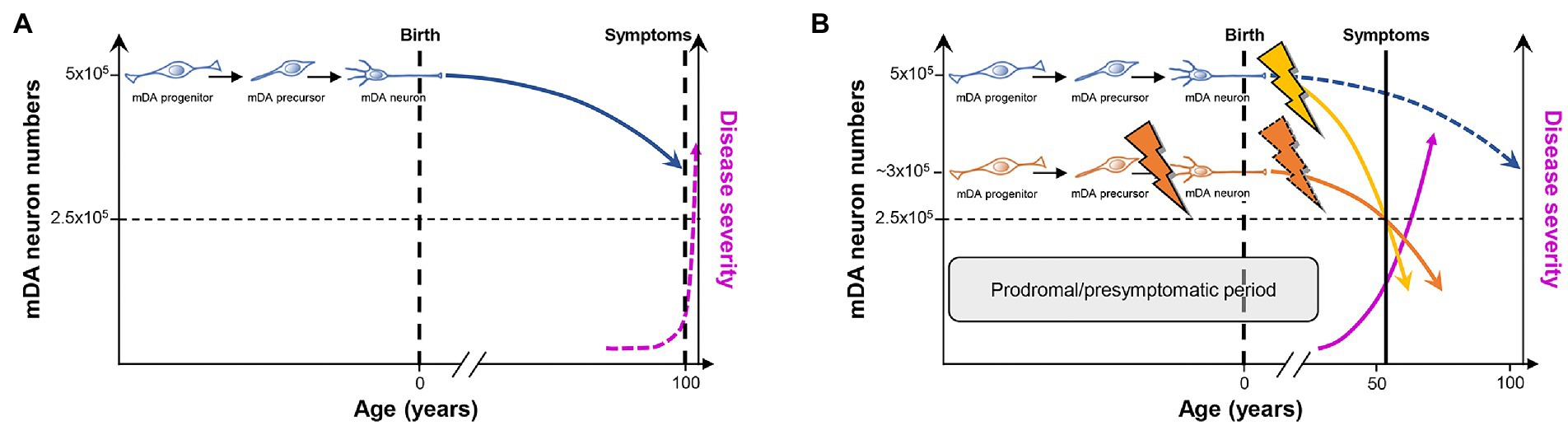
Figure 3. PD as a consequence of “multiple hits” or “stochastic acceleration” of an ongoing SNc DA neuron demise throughout the lifetime of a healthy organism. (A) Pre-and perinatal mDA neuron development (indicated by the transition from a proliferating mDA progenitor to a postmitotic mDA precursor to a maturing mDA neuron) endows a healthy human with an average number of 500,000 mDA neurons. Although still debated, a lifelong increased oxidative/nitrosative stress load coupled to a high proteostatic and energetic demand in particularly the SNc DA neurons lead to a continuous demise of some of these neurons (curved blue line) which, however, will not hit the crucial threshold (dashed horizontal black line indicating that only 50% of the mDA neurons remain in the human VM) during the usual human life expectancy. At this threshold, the first PD symptoms would appear (dashed vertical black line), and would worsen with increasing age (dashed pink line). (B) As suggested by the “multiple hit” or “stochastic acceleration” hypotheses, the normal mDA neuron demise (dashed blue line) is accelerated by the impact of diverse genetic and/or environmental stressors throughout the postnatal human lifetime (yellow flash and curved line), and will lead to the appearance of the first symptoms and diagnosis of PD (vertical black line) after reaching the critical threshold of mDA neuron numbers (dashed horizontal black line) in the sixth decade of life, as is the case in most (idiopathic) PD cases. Alternatively or in addition, a faulty pre-/perinatal mDA neuron development (orange flash) will render the individual with an already reduced number of mDA neurons at birth, which is still above the critical threshold and can thus be compensated by a heightened DA signaling and metabolism in the remaining mDA neurons from the human VM. Nevertheless, the normal age-related demise of these neurons, most likely together with additional postnatal impacts including a compromised mDA survival (dashed orange flash), will also lead to a much earlier surpassing of the critical threshold and diagnosis of PD (curved orange line). In both cases and due to the current lack of disease-modifying or-halting treatments, PD symptoms and thus severity of the disease will progressively worsen (pink line). The prodromal and presymptomatic period of life in these individuals (gray box) probably represents the best window of opportunity for the modification or, ideally, prevention of PD. Modified after (Le et al., 2009; Collier et al., 2011; Von Linstow et al., 2020).
The multiple hit or stochastic acceleration hypotheses essentially suggest that the normal, age-related decline in mDA (particularly SNc DA) neuron numbers in the healthy human brain is precipitated in the PD brain by a combination of different genetic and/or environmental factors (“hits”) impacting on inherent morphologic and/or metabolic properties of the mDA (particularly SNc DA) neurons (Carvey et al., 2006; Sulzer, 2007; Collier et al., 2011). Although the precise numbers of mDA and particularly SNc DA neurons in the adult human brain and, directly related to it, the dimension of an age-related decline in these numbers are still a matter of debate [(Collier et al., 2011; Von Linstow et al., 2020) and references therein], there is a broad consensus that the reduction of mDA neuron numbers in the aging but otherwise unaffected human brain does not reach a critical threshold required to become symptomatic during the usual human life expectancy (Collier et al., 2011; Von Linstow et al., 2020; Figure 3A). In this context, it has been suggested that mDA and in particular SNc DA neurons may have to bear a lifelong additional load (compared with other neuronal cell types) based on their particular morphology and physiology [(Sulzer and Surmeier, 2013; Surmeier et al., 2017; Surmeier, 2018; Hernandez et al., 2019; Ni and Ernst, 2022) and references therein]. Nigrostriatal (SNc) DA neurons possess long and highly branched unmyelinated axons with up to several hundred thousand synaptic sites, posing an extreme proteostatic and energetic (mitochondrial) challenge for their maintenance, and rely on an intrinsic Ca2+-based pacemaker activity and low Ca2+ buffering capacity, which also impose a high energetic (mitochondrial) demand to maintain a normal Ca2+ homeostasis in the cell (Pacelli et al., 2015). Furthermore, all DA neurons are probably exposed to an increased oxidative and nitrative stress load throughout their lifetime, simply because they synthesize and use a chemically very reactive monoamine that can be directly oxidized to quinones and free radicals (Miyazaki and Asanuma, 2008). The time-course of “normal” mDA neuron death under these already stressful but hard if not impossible to manipulate conditions might be precipitated by additional genetic (PARK and potentially also “developmental gene” mutations) and/or environmental insults, including pre-or postnatal infections and inflammation as well as environmental toxins, such as the pesticides rotenone and paraquat, the hydroxylated DA analog 6-hydroxydopamine (6-OHDA), or the accidental opioid drug by-product 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP; Carvey et al., 2006; Sulzer, 2007; Collier et al., 2011; Johnson et al., 2019; Figure 3B). These environmental toxins are, in fact, the most widely used agents to induce experimental PD in animal models of this disease, despite the reasonable questioning of this approach and its construct validity for human PD (Dawson et al., 2018). It is particularly a better control of these latter PD risk factors, including the “developmental genes,” which might provide a window of opportunity for the design of preventive measures or at least disease-modifying interventions to ameliorate the complete phenotypic emergence of this so far uncurable neurodegenerative disorder.
3. Developmental factors required for mDA neuron survival in the adult brainThe cellular and molecular underpinnings of mDA neuron development in the rodent and human brain, including the activities of gene regulatory networks and the generation of mDA neuron diversity during this process, are not discussed here and the reader is referred to excellent reviews on these topics (Arenas et al., 2015; Blaess and Ang, 2015; Bodea and Blaess, 2015; Bissonette and Roesch, 2016; Fu et al., 2016; Ásgrímsdóttir and Arenas, 2020; Poulin et al., 2020; Fiorenzano et al., 2021). The development of these neurons proceeds through a series of events in the pre- and postnatal mammalian brain. Prenatal events (during embryonic development) are the initial establishment of a VM domain capable of generating these neurons (regionalization or patterning), the proliferation of already committed mDA progenitors in this domain and their subsequent cell cycle exit to generate postmitotic mDA precursors (induction and fate specification), and the migration of these mDA precursors to their proper positions in the developing VM and at the same time acquirement of their distinctive cellular and molecular phenotypes (differentiation). The maturation of the mDA/basal ganglia system, by establishing the proper connectivity with their efferent targets (Figures 1, 2) and afferent inputs, already begins before birth but continues well into postnatal stages. All these steps are controlled by the (inter-) action of several inter- and intracellular signaling cascades, including the fibroblast growth factor (FGF; Ornitz and Itoh, 2015), sonic hedgehog (SHH; Pak and Segal, 2016; Bangs and Anderson, 2017; Petrov et al., 2017), transforming growth factor beta (TGFb)/bone morphogenetic protein (BMP; Feng and Derynck, 2005; Katagiri and Watabe, 2016), and WNT (“Wingless-type MMTV iNTegration site”; Nusse and Clevers, 2017; Wiese et al., 2018) signaling pathways (section 3.1; Figure 4), and a number of different TFs working up-or downstream of these signaling cascades or other mDA-specific genes (section 3.2; Brodski et al., 2019).
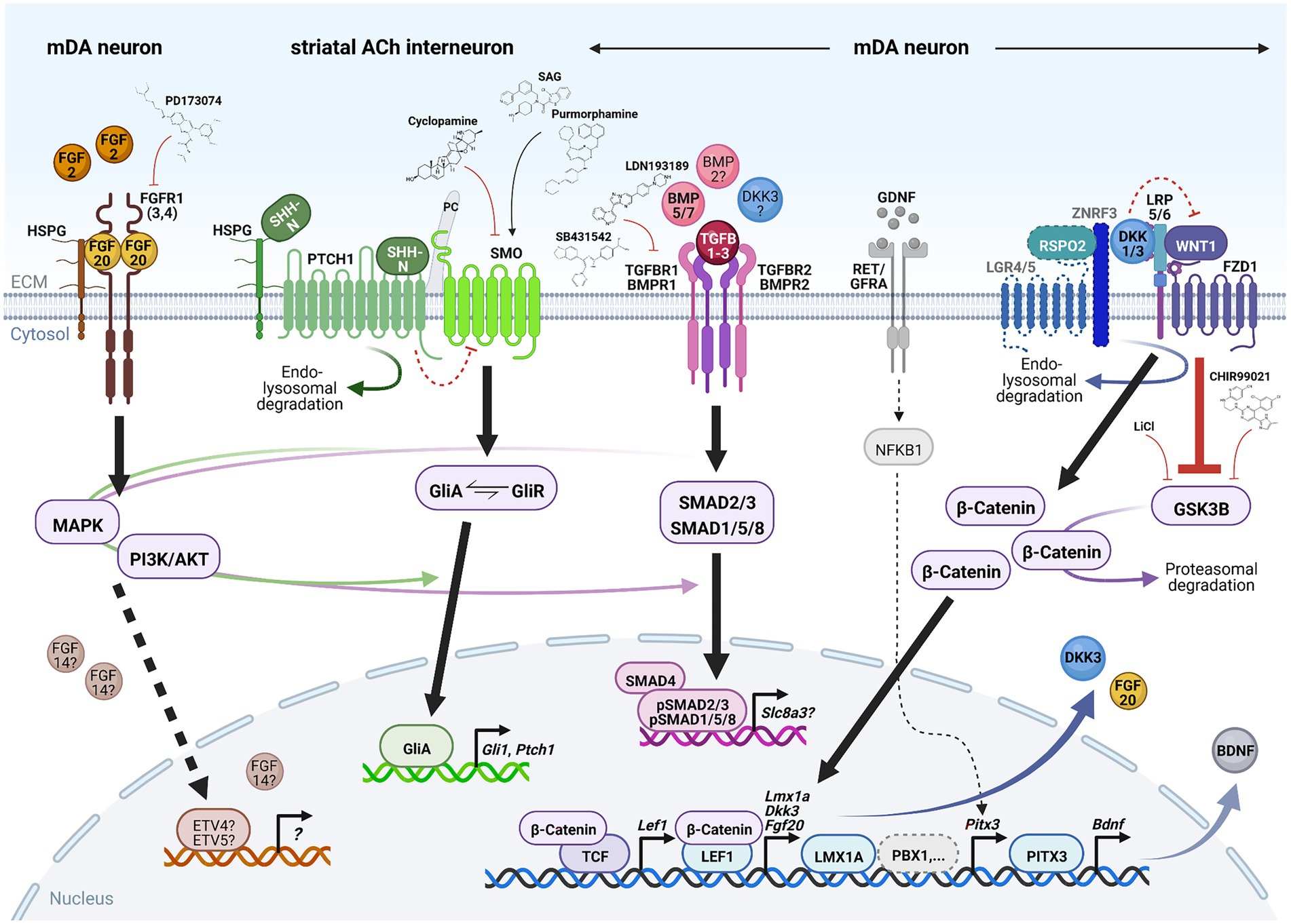
Figure 4. Signaling pathways required for mDA neuron survival in the adult, aging or lesioned mammalian VM. Simplified schematic depiction of the FGF (brown), SHH (green), TGFb/BMP (pink) and WNT/b-catenin (blue) signaling pathways and TFs or transcriptional targets implicated in the adult survival and protection of mDA neurons. FGF, TGFb/BMP and WNT/b-catenin signaling take place in the mDA neurons themselves, whereas SHH signaling is not active in these neurons but in one of their striatal target cells [cholinergic (ACh) INs]. The simplified GDNF/RET/GFRa signaling complex (gray) has been added for clarity in this context. Small molecules frequently employed to activate or inhibit the corresponding pathway are also indicated. Arrows indicate activation, red crossbars indicate inhibition of the corresponding pathway or target gene/protein. Stippled lines label pathways or signaling components not yet demonstrated in adult mDA neurons. Question marks denote still unclear target genes, TFs or ligands for the corresponding pathway during mDA neuron maintenance. See text for details and abbreviations. Created with BioRender.com.
3.1. Signaling pathways 3.1.1. FGF signalingFGFs are a large family of signaling molecules comprising 22 members grouped into seven subfamilies (Ornitz and Itoh, 2015). Five of these seven subfamilies belong to the “canonical” FGFs, including the FGF1, FGF8, and FGF9 subfamilies discussed here, which are secreted into the extracellular space and tightly bound by heparan sulfate proteoglycans (HSPGs) to limit their diffusion and to regulate their interaction with the FGF receptors (FGFRs; Ornitz and Itoh, 2015; Figure 4). The other two FGF subfamilies are either endocrine FGFs (FGF15/19 subfamily, requiring other co-factors for FGFR signaling) or intracellular FGFs (FGF11 subfamily, discussed here, without any obvious extracellular signaling activities; Ornitz and Itoh, 2015). The FGFs bind to four FGFRs (1–4) belonging to the tyrosine kinase receptor superfamily. The FGFRs consist of three extracellular immunoglobulin-like domains and two intracellular tyrosine kinase domains, which are activated after FGF ligand binding and receptor dimerization (Figure 4). The different FGFs exhibit distinct binding affinities to these FGFRs, which are additionally modulated by the existence of splice variants in particularly the extracellular FGFR domains (Ornitz and Itoh, 2015). The activated FGFRs transduce the FGF signal via four major intracellular signaling pathways, consisting mostly of sequential phosphorylation/dephosphorylation cascades: RAS/mitogen-activated protein kinase (MAPK), PI3K/AKT serine/threonine kinase, phospholipase C gamma and signal transducer and activator of transcription (Ornitz and Itoh, 2015). Ultimately, the activation of the FGF/FGFR signaling pathway leads to the regulation of gene expression in the nucleus, most prominently via the ETS variant TFs ETV4 (PEA3) and ETV5 (ERM; Ornitz and Itoh, 2015; Figure 4).
FGFs have pleiotropic functions during vertebrate development and adult homeostasis, and are thus frequently associated with human disease (Ornitz and Itoh, 2015). The most prominent FGF family members in the context of mDA neuron development and maintenance as well as PD are FGF8, FGF20, FGF2, and FGF14. FGF8, the founding member of the FGF8 subfamily, plays an important role in the early events of mDA neuron generation. These include the initial establishment of the mDA domain in the VM, the induction and proliferation of mDA progenitors in this domain, and their subsequent cell cycle exit and fate specification to generate postmitotic mDA precursors [(Harada et al., 2016; Brodski et al., 2019) and references therein]. However, Fgf8 is not expressed in the postnatal rodent brain, and the maturation and adult maintenance of the mDA neurons did not appear to depend on FGF8 function (Liu et al., 2021). Thus, other FGFs take over at these later developmental and adult stages and in PD.
The most notable member in this context is FGF20, belonging to the FGF9 subfamily of secreted FGFs (Itoh and Ohta, 2013). FGF20 was initially cloned by homology to the other two FGF9 subfamily members, FGF9 and FGF16, and shown to be highly conserved between rodents and humans (Kirikoshi et al., 2000; Ohmachi et al., 2000; Jeffers et al., 2001). In the mouse embryo, Fgf20 is widely expressed in ectodermal, mesodermal and endodermal derivatives [(Ornitz and Itoh, 2015) and references therein; (Hajihosseini and Heath, 2002)], but its expression in the developing VM has not been reported so far. Strikingly, Fgf20 transcription was almost exclusively restricted to the SNc and cerebellum in the adult rodent brain (Ohmachi et al., 2000; Jeffers et al., 2001). Subsequent in vitro analyses showed that FGF20 protein was capable of preventing mDA neuron cell death and improving their survival under serum-deprived and excitotoxic/neurotoxic conditions (Ohmachi et al., 2000, 2003; Murase and McKay, 2006). FGF20 bound to the extracellular domain of the c-isoform of FGFR1, which is one out of three (together with FGFR3 and FGFR4) and the most prominently expressed FGFR in adult rodent and human SNc DA neurons (Matsuo et al., 1994; Walker et al., 1998; Sleeman et al., 2012; Boshoff et al., 2018; Figure 4). FGF20 activated the MAPK and PI3K/AKT signaling cascades in these neurons, mediating its neuroprotective effects in vitro (Ohmachi et al., 2003; Murase and McKay, 2006; Figure 4). Notably, (Murase and McKay, 2006) also showed that FGF20 signaling via the MAPK pathway promotes TH phosphorylation at specific serine residues, thereby enhancing the activity of this enzyme and the synthesis of DA in cultured mDA and particularly SNc DA neurons (Murase and McKay, 2006). More recently and using an immunochemical detection method, (Boshoff et al., 2018) proposed that FGF20 is not expressed in SNc DA neurons but in astrocytes located in the adjacent substantia nigra pars reticulata (SNr), and might thus act in a paracrine manner on these neurons. The exact identity of the cells secreting FGF20 in the adult mammalian VM therefore remains to be established. Nevertheless, signaling via the FGFR1 and MAPK/PI3K/AKT pathways was necessary for the sustained survival of mDA neurons in the adult rodent VM, as demonstrated by a reduction of mDA neuron numbers and striatal DA innervation after intranigral viral infection or transfection of a construct encoding a tyrosine kinase-lacking FGFR1 (Corso et al., 2005), in transgenic mice expressing this dominant negative (DN) FGFR1 in catecholaminergic neurons (Klejbor et al., 2006), and after treatment with an FGFR antagonist (Boshoff et al., 2018). The strongest support for a role of FGF20 in adult mDA neuron survival, however, stems from the meanwhile numerous and reconfirmed associations of polymorphisms in the human FGF20 gene with an increased risk for PD [(Itoh and Ohta, 2013) and references therein; (Nalls et al., 2019)].
FGF2 belongs to the FGF1 subfamily lacking a signal peptide targeting them for secretion, but was nevertheless released into the extracellular space by direct translocation across the cell membrane and was also detected in the nucleus (Ornitz and Itoh, 2015). FGF2 is widely expressed in the developing rodent brain (Giordano et al., 1991), and appeared to be required for the proper establishment of the SNc DA domain during murine development, because zygotic absence (knock-out, KO) or overexpression (OE) of Fgf2 showed an inverse correlation with the numbers of SNc DA neurons, probably due to overcompensation by other FGFs (Timmer et al., 2007). FGF2 is also expressed in the adult rodent and primate (monkey and human) brain, including the SNc DA neurons and other cells (presumably glia) in the SNr (Bean et al., 1991; Cintra et al., 1991; Tooyama et al., 1994; Gonzalez et al., 1995). Subsequent in vitro and in vivo analyses corroborated a pro-survival and neuroprotective function of this growth factor in developing and mature mDA neurons [(Liu et al., 2021) and references therein]. Most importantly, the lack of Fgf2 exacerbated the SNc DA neuron loss after 6-OHDA lesioning of the corresponding null mutant mice, whereas the OE of Fgf2 in transgenic mice promoted the survival of these neurons under the same conditions (Timmer et al., 2007). However, these findings remain disputed as they were not confirmed by another group using the same Fgf2−/− KO mice but somewhat different methodological approaches (Zechel et al., 2006). Together with earlier disappointing outcomes of FGF2 infusion into the lesioned primate brain (section 4.1.1), it remains to be seen whether FGF2 may still hold up as an important factor for mDA and particularly SNc DA neuron survival in vivo (Liu et al., 2021).
In contrast to the previous FGFs, FGF14 belongs to the FGF11 subfamily of non-secreted, intracellularly localized FGFs that do not appear to interact with FGFRs (Ornitz and Itoh, 2015; Di Re et al., 2017; Figure 4). Fgf14 was identified as one potential quantitative trait locus associated with an increased number of mDA neurons and increased TH enzyme activity in the VM of inbred mouse strains (Vadasz et al., 2007). In the developing rodent embryo, Fgf14 transcription is mostly confined to the brain including the VM (Wang et al., 2000; Nouri et al., 2020), but in the adult human and mouse brain, FGF14/Fgf14 mRNA was not or barely detected in SNc DA neurons (Smallwood et al., 1996; Wang et al., 2002). However, FGF14/Fgf14 mRNA and FGF14 fusion proteins were strongly expressed within the basal ganglia circuitry including efferent output and afferent input areas of these neurons, such as the striatum (caudate putamen), globus pallidus and SNr (Wang et al., 2002). Fgf14 KO mice exhibited dyskinetic movement disturbances, which were not due to changes in mDA neuron numbers and their striatal projections or DA metabolism but due to reduced responses to DA agonists (Wang et al., 2002). This study suggested that FGF14 is implicated in mDA synaptic transmission and plasticity, potentially through its direct or indirect interaction, respectively, with voltage-gated sodium (Na+) or potassium (K+) and Ca2+ channels (Di Re et al., 2017). FGF14 might therefore act as an anterograde or retrograde intracellular modulator of DA neurotransmission in the basal ganglia system crucially involved in motor and other behavioral control (Wang et al., 2002). Several polymorphisms in the human FGF14 gene have been associated with an increased risk for neuropsychiatric disorders, including DA-related ones such as schizophrenia and SUD [(Di Re et al., 2017) and references therein].
3.1.2. SHH signalingThe SHH signal transduction pathway is probably the most complex and least understood of all four signaling pathways discussed here. SHH is one of three lipid-modified members of the hedgehog (HH) protein family, whose N-terminal bioactive fragments (SHH-N) bind to and activate the 12-pass transmembrane protein Patched 1 (PTCH1; Pak and Segal, 2016; Petrov et al., 2017; Figure 4). Binding of SHH-N to PTCH1 is aided by several co-receptors (CDON, BOC, GAS1) necessary for HH pathway activation (Pak and Segal, 2016; Petrov et al., 2017). In the absence of SHH, PTCH1 represses the activity of the seven-pass transmembrane protein Smoothened (SMO) by a not yet understood mechanism (Figure 4). Binding of the SHH-N ligand to the PTCH1 receptor causes the endocytosis and lysosomal degradation of this ligand-receptor complex and the relocation of SMO from an intracellular compartment to the primary cilium (PC), where PTCH1 is normally located (Figure 4). Subsequently, SMO and the downstream effectors of this pathway, the GLI zinc finger TFs comprising three members in vertebrates (GLI1-3), are activated by a complex sequence of protein kinase-, G-protein-coupled receptor kinase- and phosphatase-mediated phosphorylation and dephosphorylation events (Pak and Segal, 2016; Petrov et al., 2017). Gli1 is a direct target gene of the HH signaling pathway and thus positive feedback regulator of this pathway, whereas GLI2 and GLI3 function mostly as transcriptional activator (GliA) or repressor (GliR), respectively (Pak and Segal, 2016). Phosphorylation of the full-length GLI proteins (GliA) targets them for proteolytic cleavage and proteasomal degradation, which in the first case yields the GliR form of the corresponding protein. The signaling outcome of the HH pathway is determined by the balance between the GliA and GliR forms of these TFs and their control of the transcriptional output in the nucleus, including positive and negative feedback regulation by their targets Gli1 and Ptch1, respectively (Pak and Segal, 2016; Figure 4). The PC plays a fundamental role in HH signal transduction, as evidenced by the disruption of this pathway in mouse mutants for several structural and functional components of the PC (Bangs and Anderson, 2017). Not only PTCH1 and SMO have to localize to the PC for proper HH signaling but also the GLI TFs, which are bound as full-length (GliA) proteins by SUFU, a negative regulator of this pathway, and translocated into the tip of the PC. In the PC, GliA proteins will either serve as a “reserve pool” for HH signal transduction or become phosphorylated and processed into the GliR form (Pak and Segal, 2016; Bangs and Anderson, 2017). The PC also plays a role in other signal transduction pathways, such as WNT/b-catenin and TGFb signaling (Ma et al., 2022).
SHH is the sole HH protein in the developing and adult mammalian brain, and has therefore been implicated in a variety of neurodevelopmental processes and neurodegenerative diseases (Brodski et al., 2019; Yang et al., 2021). In the developing mouse VM, SHH signaling is required only for the ventral patterning and establishment of the mDA progenitor domain, but not for mDA neuron differentiation. This is due to the temporally limited expression until midgestation of essential SHH signaling components (PTCH1, GLI1/2/3 and SHH itself) in this region [(Brodski et al., 2019) and references therein]. Some components of SHH signaling, such as SMO, continue to be expressed in maturing mDA neurons and control the proper axonal outgrowth of a subset of these neurons (Brodski et al., 2019). The conditional inactivation (cKO) of structural and functional components of the PC in mice also resulted in a partial or transient reduction of mDA progenitors and neurons, depending on the developmental timepoint at which SHH signaling was affected in these cKO mice [(Brodski et al., 2019) and references therein].
Despite the limited role of SHH signaling during mDA neuron development, the improved mDA neuron survival after intrastriatal or supranigral injection of recombinant human (rh) lipid-modified SHH-N protein were among the first reports suggesting a neuroprotective role of this signaling pathway in adult mDA neurons (Dass et al., 2002; Tsuboi and Shults, 2002). Two seminal studies by Gonzalez-Reyes et al. (2012) and Ortega-de San Luis et al. (2018) later demonstrated that SHH is expressed by virtually all mDA neurons (including the SNc) in the adult mouse brain, although essential components of this pathway, such as the PTCH1/2 receptors, were not detected in these neurons. Moreover, Smo cKO in dopamine transporter (DAT/SLC6A3)-expressing (maturing) mDA neurons did not result in any overt mDA phenotype up to 18 months of age, even after a neurotoxic (6-OHDA) challenge (Zhou et al., 2016). The hyperactive locomotor phenotype, attenuated psychostimulant response, and reduced Bdnf but unchanged Gdnf and Bmp7 transcription in the VM of young adult Dat-Smo cKO mice observed by Zhou et al. (2016) might thus be due to the known function of SMO as an axonal guidance receptor in mDA neurons (Hammond et al., 2009). Dat-Shh cKO mice did not display an obvious mDA phenotype at birth, but revealed a progressive 40% loss of in particular the SNc DA neurons starting at 4 months and plateauing in the 8 month of age (Gonzalez-Reyes et al., 2012). These losses were accompanied by highly dynamic changes in striatal mDA innervation and DA release, locomotion as well as in the expression of other proteins involved in DA neurotransmission, such as TH, DAT, DRD2 and the vesicular monoamine transporter VMAT2 (SLC18A2), suggesting temporary compensatory mechanisms in these mouse mutants (Gonzalez-Reyes et al., 2012). As a cautionary tale, the behavioral and gene expression phenotypes of the Dat-Smo and Dat-Shh cKO mice might have also been caused, at least in part, by the disruption of one Dat allele in the corresponding Cre driver mice (Giros et al., 1996; Zhuang et al., 2005; Backman et al., 2006). The absence of the PTCH1 receptor in adult mDA neurons prompted the investigation of a non-cell autonomous (paracrine) neuroprotective action of SHH within the nigrostriatal pathway. Indeed, mDA neuron death was paralleled by a progressive loss of cholinergic and GABAergic FS INs in the Dat-Shh cKO striatum (Gonzalez-Reyes et al., 2012). This finding was later refined by (Ortega-de San Luis et al., 2018) using more sophisticated methods, showing that the physiologically induced loss of more than 95% mDA neurons resulted in a selective reduction of only GABAergic FS INs but not cholinergic INs and, conversely, Smo cKO in these two striatal IN populations affected the adult survival of only the cholinergic but not the GABAergic FS INs. In search for a non-mDA source of SHH in the striatum, the GABAergic FS and cholinergic INs themselves were identified as potential auto- or paracrine suppliers of this signaling molecule (Ortega-de San Luis et al., 2018). Although Gonzalez-Reyes et al. (2012) proposed that Gdnf transcription in these striatal INs, one of the most potent neurotrophic factors for mDA neurons (Chmielarz and Saarma, 2020), was engaged in a mutual negative feedback loop with the transcription of Shh in mDA neurons, some of their findings were not reproduced by Ortega-de San Luis et al. (2018). Adult Smo cKO mice lacking an active SHH pathway in the cholinergic INs showed a modest but progressive loss of mDA neurons which, however, was not due to changes in striatal GDNF expression. These data revealed that the mDA neurons and part of their striatal target cells are engaged in an intricate homeostatic relationship whose details still remain to be clarified. All three neuronal populations require the trophic support of a factor produced by either one of the other neuronal population or by themselves: striatal cholinergic INs require SHH produced by the mDA neurons or released locally; striatal GABAergic FS INs require mDA input independent of SHH; and mDA neurons require an unknown cholinergic IN-derived trophic factor for their survival.
Striatal cholinergic INs are also thought to play a crucial role in the emergence of L-DOPA induced dyskinesias (LIDs), a severe side-effect of prolonged L-DOPA medication (Shen et al., 2022). The potential role of SHH signaling in the regulation of striatal cholinergic IN physiology prompted (Malave et al., 2021) to test whether the acute or chronic co-application of a SHH agonist (SAG or purmorphamine) or antagonist (cyclopamine) during L-DOPA treatment would attenuate or increase, respectively, the appearance of LIDs in mouse and primate PD models. This was indeed the case and traced back to the inactivation or activation, respectively, of the MAPK pathway downstream of SMO in these INs (Malave et al., 2021; Figure 4). Smo cKO in striatal cholinergic INs or Shh cKO in young mDA neurons resulted in the progressive worsening of LIDs despite the lack of an mDA phenotype in these mice, whereas the OE of a constitutively active SMO protein in a genetic PD mouse model blocked the appearance and intensification of LIDs despite the severe mDA phenotype of these animals (Malave et al., 2021). In an elegant series of experiments, Malave et al. (2021) showed that the repeated optogenetic stimulation of mDA burst firing led to LID-like behavior intensification in an L-DOPA-independent manner. This was most likely due to the exhaustion of SHH signaling from the mDA neurons to the striatal cholinergic INs, because a single dose of the antagonist cyclopamine prior to stimulation onset exacerbated, whereas the agonist SAG blocked this effect. Importantly, these findings suggest that an imbalance between excessive DA signaling (due to L-DOPA administration) and strongly diminished SHH signaling (due to mDA neurodegeneration) in striatal cholinergic INs leads to the emergence of LIDs. The co-administration of SHH agonists might restore this balance in the L-DOPA treated PD brain and provide an effective or even preventive anti-dyskinetic treatment.
The importance of the PC in nigrostriatal DA neurotransmission and survival has only been recognized recently. PC morphology (length) in the striatal targets (MSN and IN) of the mDA neurons was regulated by DA-mediated (on the presynaptic side) and DRD2-specific (on the postsynaptic side) neurotransmission, leading to elongated PC in their absence (Miyoshi et al., 2014). Elongated PC were also detected in postmortem striatal neurons of iPD patients and unilaterally treated 6-OHDA mice, suggesting the loss of mDA innervation as the causative factor in both cases (Schmidt et al., 2022). PC formation was reduced specifically in striatal cholinergic INs and astrocytes of humanized mice carrying autosomal dominant mutations (R1441G and G2019S) in the human LRRK2/PARK8 gene (Dhekne et al., 2018; Khan et al., 2021). These LRRK2 mutations are thought to increase the kinase activity of the mutant proteins, and were detected in late-onset fPD (Vázquez-Vélez and Zoghbi, 2021). Lrrk2 is highly expressed in the adult mouse striatum (Benson and Huntley, 2019). The increased phosphorylation of essential PC components by the mutant LRRK2 proteins, although not directly demonstrated in striatal cells, interfered with PC formation and disrupted SHH signaling in LRRK2 R1441G mouse embryonic fibroblasts and striatal cholinergic INs as well as LRRK2 G2019S human induced pluripotent stem cells (hiPSCs; Dhekne et al., 2018; Khan et al., 2021). This suggested that the survival of striatal cholinergic INs and, even more importantly, the intricate SHH-mediated nigrostriatal homeostatic feedback loop might be compromised in the LRRK2 mutant PD brain. PC are also present in a substantial proportion of adult SNc DA neurons (non-SHH-responsive, see above). Their enhanced biogenesis and elongation under increased mitochondrial oxidative stress conditions (MPTP lesioning) appeared to exert a neuroprotective effect by augmenting the autophagic removal of dysfunctional mitochondria and suppressing the apoptotic death of these neurons (Bae et al., 2019). These results were partly corroborated by a recent study showing that the cKO of a crucial structural and functional component of the PC in maturing mDA neurons (Dat-Ift88 mice) resulted in the loss of approximately 20% SNc DA neurons and a corresponding decrease of striatal mDA innervation and DA content, as well as an increase of PC length in striatal neurons (Mustafa et al., 2021). The reduction of SNc DA neuron numbers in the Dat-Ift88 cKO mice was not further accentuated by MPTP treatment (Mustafa et al., 2021), in contrast to the report by Bae et al. (2019). The most likely explanation for these discrepancies are methodological differences (constitutive genetic ablation vs. viral OE-mediated knockdown of the PC component, and subchronic vs. acute administration of MPTP) between the two studies (Bae et al., 2019; Mustafa et al., 2021). Mustafa et al. (2021) also proposed that the loss of SNc DA neurons in the Dat-Ift88 cKO mutants affected primarily a PC-bearing subset of these neurons displaying a high intrinsic electrophysiological activity that can be further stimulated by DA, which might therefore be particularly prone to metabolic stress and degeneration. Proliferating neural precursors and maturing mDA neurons derived from iPD and fPD hiPSCs, considered as “rejuvenated” cells representing fetal stages in human development, were recently reported to display mitochondrial respiratory deficits, dysregulated pathways associated with PC and SHH signaling, and shortened PC (Schmidt et al., 2022). These phenotypes were reversed by the application of cyclopamine, a SHH antagonist directly binding to SMO, or the withdrawal of purmorphamine (a SMO agonist; Figure 4) from the culture media, and thus interpreted as a consequence of increased SHH signaling in the iPD- and fPD-derived neural cells (Schmidt et al., 2022). The mitochondrial impairments in these “rejuvenated” cells support the assumption of a developmental onset of metabolic deficits in PD (discussed in section 5), although it remains to be clarified how the proposed SHH signaling impairments in the iPD/fPD mDA precursors and neurons can be reconciled with the absence of the PTCH1/2 receptors and GLI TFs in these cells.
3.1.3. TGFbeta/BMP signalingLong known as secreted neurotrophic proteins, the 33 members of the TGFb superfamily in mammals are grouped in two major subfamilies based on their sequence and structural homology: the TGFb-like subfamily, comprising the three TGFB1-3 isoforms (discussed here), activins, nodal and some growth and differentiation factors (GDFs); and the BMP-like subfamily, consisting of 13 BMPs (discussed here) and most GDFs (Weiss and Attisano, 2013). The bioactive peptides of this superfamily are cleaved from larger precursor proteins and dimerized by the formation of disulfide bonds. After release, their activity in the extracellular space is controlled by the interactions with the extracellular mat
留言 (0)