

記住我





Metabolic disorders, particularly obesity increases the risk of a number of cancers, e.g. colon, mammary, pancreas, liver (1, 2), etc. Obesity, which occurs in half of the US population, is now recognized as a confounding factor for cancer-related death (3, 4). The contribution of lipid dysfunction to cancer is particularly high for liver cancer. The mortality risk for liver cancer is estimated to be 4.52-fold higher in men with >35 body mass index (BMI) compared with those with BMI <29 (1). Liver steatosis is a common comorbid disease for liver cancer and is associated with metabolic diseases including obesity, insulin resistance (IR), and diabetes as well as in other related disorders such as alcohol usage disorders (5). While hyperinsulinemia, hyperglycemia, and hyperlipidemia as a result of peripheral insulin resistance and metabolic disorder can directly contribute factors to promote tumorigenesis (6), the resulting development of liver steatosis due to these conditions directly establishes the microenvironment to promote tumor development. This review will focus on the local tumor microenvironment in liver steatosis for its role in promoting cancer development.
Contribution of steatosis to liver cancerIn the liver, steatosis is defined when at least 5% of lipid droplets are accumulated among hepatocytes in the histopathological diagnosis (7, 8), and is classified as alcoholic or nonalcoholic forms due to etiology. Non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) develop in patients with metabolic syndromes including obesity, IR and diabetes (9, 10) whereas alcoholic liver disease (ALD) and ASH (alcoholic steatohepatitis) are caused by excessive alcohol drinking which also contributes to lipid metabolic dysfunction (11, 12). While simple fatty liver is reversible by lifestyle changes, ASH and NASH can progress to more morbid forms of liver pathologies including fibrosis/cirrhosis and is highly associated with liver cancer (13).
Patients with varying degrees of steatosis are susceptible to hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA), the two dominant forms of liver cancer. In particular, the NAFLD-HCC incidence ratio increased significantly (1.92-fold for men and 12.7-fold for women) in the last 20 years whereas it decreased or remain unchanged for many other major etiologies of HCC (14). This increase is concurrent with the increase of obesity epidemic particularly in women, suggesting a role of lipid dysfunction in liver carcinogenesis. Consistently, alcohol consumption and associated alcoholic liver disease was estimated to be an independent risk factor for poor disease-free survival, particularly in non-virus hepatitis associated HCC (15).
Earlier studies using chemical carcinogen to induce cancer formation found that high fat diet (HFD) feeding significantly induced cell proliferation in diethyl nitrosamine (DEN) induced HCC models (16). While this observation is supported by high fructose, high cholesterol and alcohol feeding studies (17–19), other experiments show that HFD protects against DEN induced liver injury, leading to reduced HCC (20, 21). Avoiding the complications of chemical induced injury, genetic models were used to explore liver cancer development. Liver cancer is highly heterogenous on the pathohistological levels as well as genetic landscape. In recent years, exome sequencing has led to the discovery of TERT, CTNNB1 and TP53 as the dominant mutations and PI3K/AKT/PTEN/mTOR together with MAPK pathway as the primary signaling pathways that promote liver cancer development together with Wnt/β-catenin signaling pathway (22, 23). Mutation of TERT1 promoter is found to be a primary characteristic of NAFLD associated liver cancer (24) and loss of telomerase promotes metabolic dysfunctions in hepatocytes (25). Activating mutation of CTNNB1 (encodes β-catenin) occurring in 37% of HCC is thought to support the growth and transformation of liver cancer stem cells (26–29). As such, activating mutation of CTNNB1 confers the oncogenic potential of β-catenin and promotes HCC development (26, 27). Interestingly, manipulation of neither TERT, CTNNB1 nor TP53 by themselves is sufficient to result in liver tumor development (25, 26, 30, 31). Activation of PI3K signaling pathway, however unequivocally resulted in the development of HCC and CCA. Activating PI3K/AKT signal via deletion of Pten showed spontaneous tumor development following steatosis and fibrosis (32–35). The PI3K/AKT signal upregulation results in increased lipid anabolic metabolism in addition to acting as a pro-growth and pro-survival signal (35–45). In the Pten deletion model, inhibiting steatosis attenuates or abolishes tumor development, suggesting that steatosis is required for liver tumor development (32, 33), whereas short term feeding of HFD accelerates the development of tumors (46). The PI3K/AKT signal is necessary for driving the steatosis phenotypes in the liver (35, 37). As such, introduction of activated AKT delivered through hydrodynamic injection of myristylated AKT is necessary to drive the development of HCC and CCA for a number of signals including Notch, YAP, Shp2, Hippo and others (28, 47–49). Consistent with this notion, combining other genetic models with non-genotoxic chemicals and diet manipulations demonstrated that liver injury and steatosis promotes the development of tumors (28, 31–33, 50–52). In several mouse models including those lacking p53 and Indian Hedgehog, consumption of a Western-style diet, or a high-fat/high-cholesterol diet to the point of developing hepatic steatosis was shown to promote higher liver tumor incidence than the control diet group (53, 54).
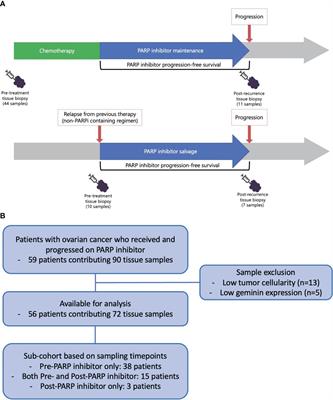
In these genetic models where HFD feeding accelerates/promotes tumorigenesis, liver injury is a main consequence associated with steatosis (33, 46, 55). In fact, the effect of p53 on hepatocytes apoptosis may have contributed to the lack of tumorigenic effects observed in p53 deletion mice (31, 51) as p53 deficiency protected hepatocytes from undergoing apoptosis in response to HFD feeding and subsequent liver injury (56). Similarly, while activated β-catenin mutation is capable of promoting hepatocyte regeneration, the genotoxic effect requires steatosis and/or liver injury to promote liver cancer development (26, 28, 30). In fact, the function of β-catenin in sustaining normal hepatocyte function explains how β-catenin loss also promotes a protumor environment (57, 58). Deletion of β-catenin leads to loss of liver zonation, resulting in spontaneous repopulation of β-catenin+ cells due to hepatocyte death associated with loss of zonation. The death of hepatocytes also leads to cancer development from the β-catenin+ cells when genotoxic chemicals are introduced (29, 58, 59). Similar to chemical induced injury, steatotic injury has been shown to induce Wnt signal in the liver and elsewhere (60–63). Together, these studies suggest that liver steatosis establishes a microenvironment that promotes the growth of liver cancer cells and permits the expansion of any initial genotoxic events to develop into tumors (Figure 1).
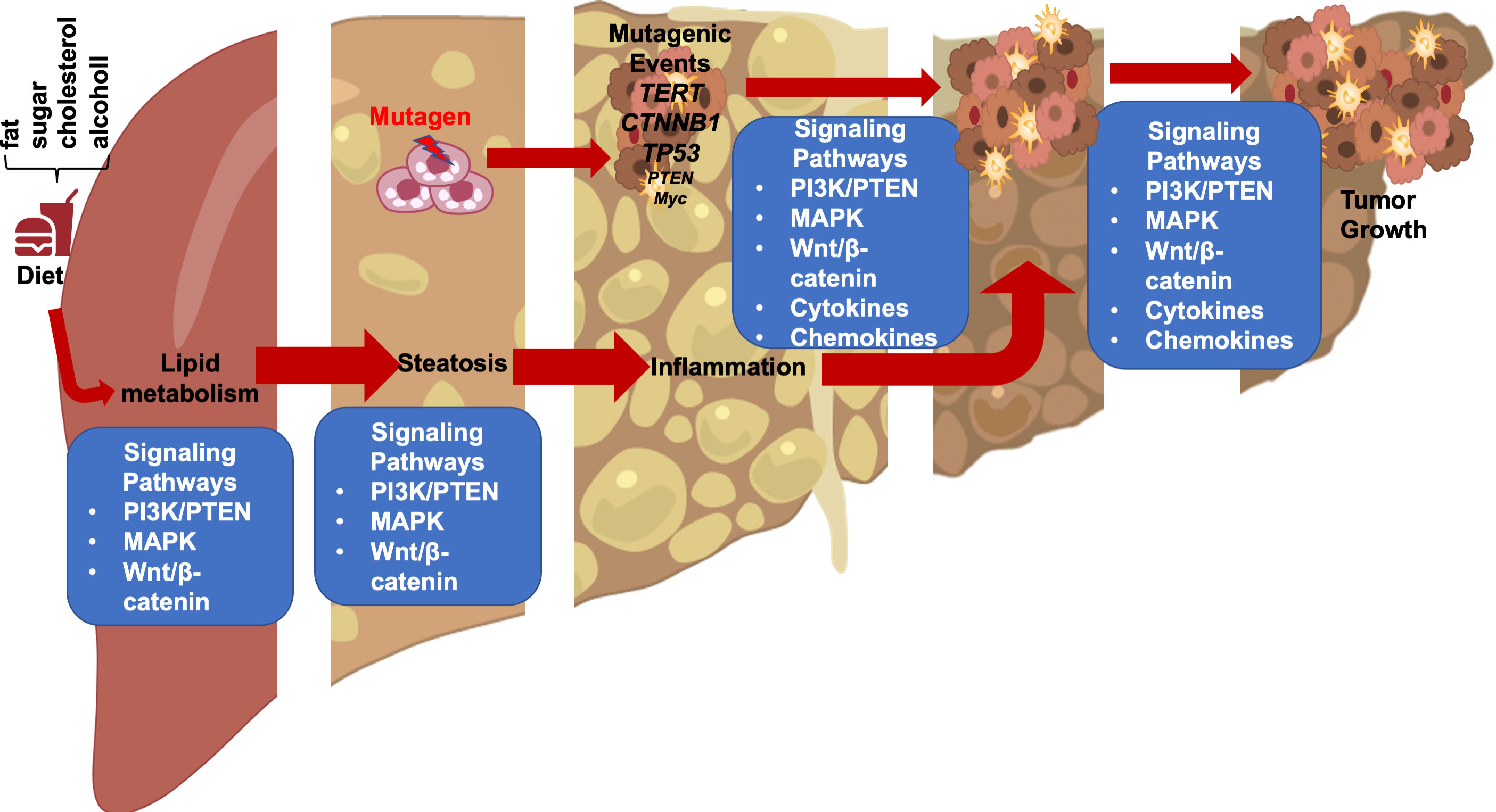
FIGURE 1
Figure 1 Steatotic Liver Damage Establishes a Tumor Microenvironment. The primary functions of the liver are metabolism and detoxication. Nutrients from the gut are metabolized in the liver involving the insulin regulated PI3K/PTEN pathway. Wnt/β-catenin signaling also plays important role in regulating the metabolic and detoxicating functions of the liver as it regulates liver structure and zonation. Following a diet containing high fat, sugar, cholesterol, or alcohol, activation of these signals results steatosis. The consequence cell death due to steatosis and loss of liver structure leads to inflammatory cell infiltration. Inflammatory mediators produced due to liver inflammation propagate any genotoxic events as the induce the proliferation of tumor initiating cells that carry mutations of TERT, CTNNB1, TP53 and to a lesser extend PTEN and MYC as well as others. The Wnt/β-catenin, PI3K/PTEN and MAPK signaling pathways as well as cytokine and chemokine are all implicated in the proliferation of the tumor cells and play roles in propagating the initial mutagenic events.
Macrophage response to steatotic liver injury: A double-edged sword in liver carcinogenesisThe liver is known as an immunosuppressive organ as illustrated by the lower dose of immunosuppressive therapy needed for liver transplantation as compared with other organ transplantations (64, 65). Liver macrophages play a critical role in this process. There are 2 basic populations of macrophages in the liver, the local proliferating Kupffer cells and the infiltrating monocyte derived macrophages (66). Kupffer cells are located within the liver sinusoids and play surveillance functions by monitoring pathogens coming into the liver. Being the largest tissue resident macrophage population in the body, Kupffer cells are the first responders in liver immune system. Unlike monocyte derived macrophages, Kupffer cells are highly effective in binding and clearing Escherichia coli (E. coli) brought in via the portal circulation (67).
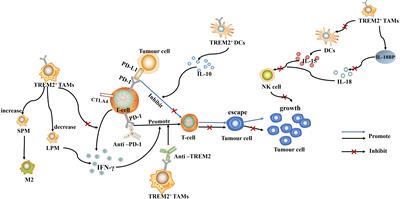
During homeostasis, Kupffer cells, but not monocyte derived macrophages, present antigens to induce immune tolerance through expansion of select regulatory T-cells and inhibition of T cytotoxic lymphocytes and to induce apoptosis in other T-cells (64, 68). In response to inflammation that cannot be cleared by Kupffer cells alone including those induced by pathogens and injury, inflammatory mediators released by Kupffer cells also recruit other inflammatory cells including monocytes-derived macrophages and neutrophils in addition to subsets of CD4+ and CD8+ T lymphocytes and NK/NKT cells (Figure 2). In particular, neutrophils, being the most abundant leukocytes in circulation are the first responders to acute inflammation to clear pathogens and damaged/dying cells. Similar to macrophages, neutrophils are highly enriched in the steatotic livers and depletion of neutrophils protects mice from experimentally induced steatohepatitis (69). Together, these infiltrating immune cells crosstalk with macrophages to clear pathogens and damaged tissues. In chronic injury conditions such as those presented by ALD and NAFLD, these inflammatory cells establish an environment that is pro-tissue repair and returning to homeostasis on the one hand; and pro-tumor growth when genotoxic events are present on the other. Recognition of damaged hepatocyte-released molecules by macrophages is important in the propagation of the signals and the sustained inflammatory response (see later section). Interaction of macrophages with other cell types such as cholangiocytes and hepatic stellate cells are also important in the disease progression and the establishment of the tumor microenvironment. This review focuses on the role of macrophages and macrophage produced inflammatory mediators. The interactions of macrophages with other inflammatory cells and their function in the tumor immune environment is also important for liver cancer development (70).
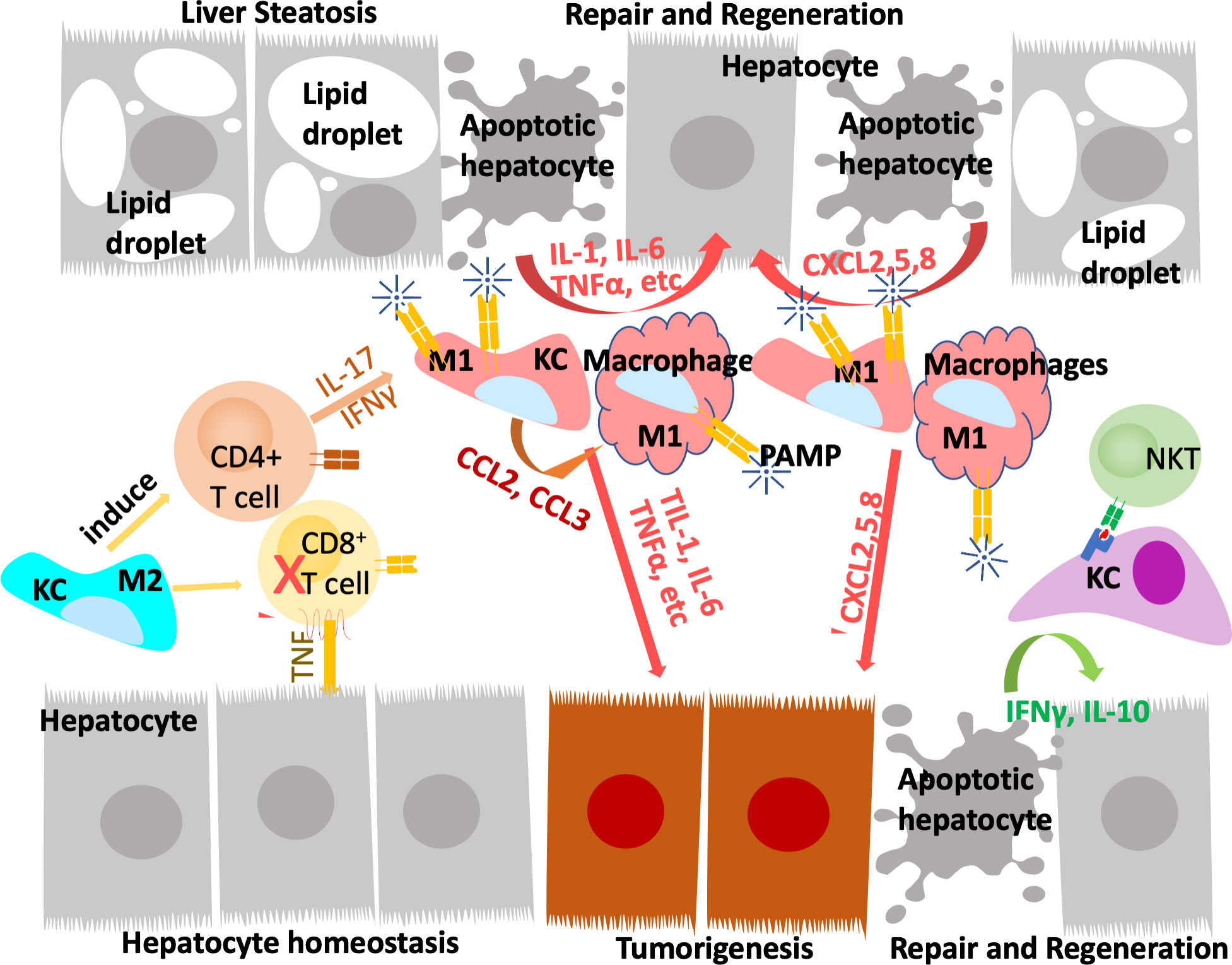
FIGURE 2
Figure 2 Innate immune system regulate liver repair and tumorigenesis due to steatosis. The liver developed a unique immune response system that tolerate gut bacterial-induced inflammation while eliminating them at the same time. During hepatocyte homeostasis (grey cells), Kupffer cells (KC) binds to and eliminate gut bacterial while producing anti-inflammatory cytokines to inhibit CD8+ cytotoxic T cells and induce their apoptosis. At the same time, Kupffer cells also induce antigen specific CD4+ Treg cells to assume tissue repair functions. In response to chronic injury presented in NAFLD and liver steatosis (grey cells with lipid droplets), Interleukine-17 (IL-17) and interferon g (IFNγ) produce from Tregs recruit monocyte derived macrophages as well as activating a M1 proinflammatory program in Kupffer cells. The proinflammatory cytokines produced by these M1 macrophages/Kupffer cells including IL-1, IL-6, TNFα, etc induces hepatocyte proliferation to repair the damaged tissues and replace apoptotic hepatocytes due to steatosis. These proinflammatory cytokines also establishes a pro-tumor microenvironment as they propagate any genotoxic events that are present in the tumor initiating cells (orange cells). The M1 macrophage/Kupffer cells also produces chemokines to promote hepatocyte and tumor cell proliferation, leading to tissue repair and/or tumorigenesis.
Steatotic liver damage establishes a pro-inflammatory tissue microenvironmentEarly studies showed that administration of liver toxicants such as carbon tetrachloride (CCL4) and 3,5-Diethoxycarbonyl-1,4-Dihydrocollidine (DDC) provoke the growth and infiltration of macrophages in the liver (71, 72). In patient samples, macrophages have been observed to be recruited to the NASH livers (73). These macrophages play roles in the disease progression of steatosis by producing inflammatory factors that sustain injury (6, 65, 74). In B6 mice fed HFD to induce NAFLD, infiltration of immature macrophages that are CD11b+Ly6ChiLy6G- are observed. These macrophages are more readily able to produce proinflammatory cytokines than those from the lean mice controls (75). In mice fed methionine-choline deficient (MCD) diet to induce NASH, induction of macrophage proinflammatory genes is found to associate with more progressive fibrosis (76). Depletion of macrophages using liposomes to deliver clodronate led to reduced expression of proinflammatory genes and attenuated the progression to NASH and fibrosis in mouse models (32, 77). In genetic models where cytokine signals are manipulated, infiltration of macrophages are also found to prolong liver injury (78). Deletion of Ccl2 (C-C motif chemokine ligands 2), a chemokine that recruits monocytes to the liver, results in reduced liver damage and fibrosis (76). Treatment with a dual antagonist for CCR2 and CCR5, receptors for CCL2 and CCL5, significantly reduced macrophages and protected rats from liver injury in a diet induced NASH model (79) and has shown promising effects for NASH in phase II clinical trial (80). Inhibiting activation of Kupffer cells and infiltration of monocytes by deletion of proinflammatory receptor Trem-1 also significantly attenuated liver inflammation, injury and liver fibrosis induced by CCL4 treatment (81). Adoptive transfer of Trem1-sufficient Kupffer cells led to reactivated inflammation and injury, suggesting that the presence of Trem1-sufficient Kupffer cells can sustain chronic inflammation. In both CCL4 induced liver injury and MCD feeding induced NASH mice, pharmacological inhibition, or deficiency of monocyte chemoattractant protein (MCP-1 or CCL2) led to reduced liver injury and inflammation (76, 82). Together, inflammation is thought to be a crucial phase for the disease progression of NAFLD and the role of macrophages appear to be important in this progression.
Steatosis induced inflammation establishes a pro-tumor microenvironmentChronic injury and the associated inflammatory responses are a major link between liver steatosis and cancer development. The development of liver cancer is a slow process that evolves from premalignant lesions developed within chronically damaged livers (83). In chemical induced hepatocarcinogenesis, HFD feeding promotes hepatic inflammation and exacerbates tumor development (84). In HCC mice induced by transgenic expression of hepatitis C virus core protein, HFD feeding to induce liver steatosis significantly increased tumor incidence (85). In these mice, the toll-like receptor (TLR) signal involved in innate immune response was found to promote the transformation of liver tumor initiating cells (86). In mice lacking p53 and concurrent expression of c-Myc, T cell mediated immune surveillance was found to reduce tumor formation and increase survival. This tumor surveillance is overcome when the β-catenin pathway is induced by exogenous expression of active β-catenin, further confirming that β-catenin signal sustains tumor growth (87). In the Pten deletion model, steatosis is required for tumor growth and is accompanied by inflammation and induction of β-catenin (33, 88). It was discovered that depletion of macrophages reduces Wnt/β-catenin signals and attenuates tumor growth (32, 89). Together, these studies suggest steatosis establishes an inflammatory environment that is pro-tumor growth.
Infiltration and reprogramming of macrophages are observed in essentially all experimental models and HCC patients. In HFD fed mice where tumors are initiated by DEN treatment, macrophage recruitment accompanied chronic liver injury and liver cancer development (84). In genetic models of NAFLD-NASH-liver cancer, macrophages also play a dominant role in promoting liver cancer development. Depletion of macrophages resulted in reduced tumor incidence in the Pten deletion mice (32) and this was thought to involve TLR signaling (90). Together, this evidence suggests that while macrophages can produce pro-repair cytokines, sustained presence of macrophages can prolong liver injury and result in further liver damage.
During liver repair in response to injury, liver macrophages, particularly Kupffer cells are credited in producing pro-mitogenic cytokines to induce the growth of liver progenitor cells and promote liver regeneration (91–93). Depletion of macrophages attenuates tissue repair and resulted in exacerbated fibrogenic phenotype (92) and also led to delayed recovery of metabolic functions performed by the liver (94). When inflammation is not resolved, the signals produced by macrophages exacerbate liver injury and lead to chronic inflammatory conditions and sustain the production of proinflammatory cytokines (6, 65). During liver tumorigenesis, the chronic inflammatory condition and proinflammatory cytokines promote tumorigenesis by providing the tumor microenvironment as well as signaling the growth and promoting the proliferation of tumor initiating cells (91). High fat diet feeding induces macrophage production of a number of inflammatory factors and cytokines including interleukins, C-C ligands (CCLs), interferon γ (IFNγ) and tumor necrosis factor α (TNFα) to facilitate hepatocyte proliferation (84). Cytokines produced by these resident as well as infiltrating macrophages such as TNFα, transforming growth factor β (TGF-β), interleukin 6 (IL-6) and 18 (IL-18) are highly associated with the development and progression of hepatocellular carcinoma (HCC). In a mouse tumor model established by subcutaneous transfer of DEN-initiated liver tumor initiating cells, depletion of macrophages attenuated the progenitor cell properties and reduced tumor development (95). The presence of macrophage-produced TNFα also triggers chromosomal instability in liver tumor initiating cells, permitting propagation of genotoxic events leading to tumorigenesis (95). TNFα produced by macrophages are also proposed to promote the proliferation of liver cancer initiating cells (96). These tumor initiating cells were found to display similar transcriptome profiles as the ov-6 positive liver progenitors that express LIN-28 (97). The expression of LIN-28 allows these cells to respond to the interleukin-6 (IL-6) signal to proliferate. In MCD diet fed mice, macrophage reprograming also contributed to the proliferation of liver progenitors and promoted HCC proliferation (98). In tumors induced by expression of Myc and deletion of TP53, upregulation of β-catenin promoted immune escape of the tumors involving defective recruitment of myeloid lineage cells that include macrophages (87). As a potential driver mutation gene, activation of β-catenin is associated with liver tumor initiation (27, 48). In the Pten deleted NAFLD-NASH-Tumor mice, β-catenin was found necessary to sustain the growth of liver tumor initiating cells as deletion of β-catenin attenuated their growth (32, 33, 88). Depletion of macrophages suppressed Wnt/β-catenin signal and led to reduced tumor burden in these mice. TLR4 was found to play a role in the macrophage-promoted proliferation of tumor initiating cells and tumorigenesis in these mice (90). Together, these data suggest that macrophages may be necessary to both sustain tumor initiating cell proliferation as well as establishing the liver injury environment that allows the tumors to grow.
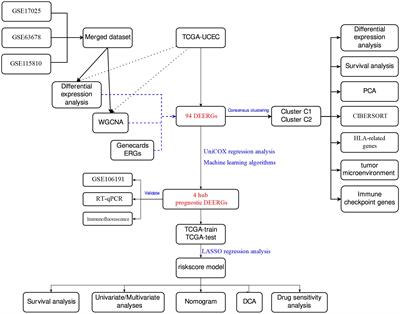
Cytokines in steatosis driven HCCInflammatory cytokines play key roles in the communication between macrophages with surrounding cell types and also reprograming macrophages to different spectrums of polarizations under given stimulatory conditions, resulting in high heterogeneity of liver macrophages (99). Beyond proliferation of resident Kupffer cells and infiltration of monocyte-derived macrophages, hepatic macrophages are also stimulated or “reprogrammed” to produce a variety of pro- and anti-inflammatory cytokines that classify them on the spectrums of M1 vs. M2 polarization (Figure 3). During steatosis driven liver cancer development, a complex interaction of anti- and pro-inflammatory cytokines promotes cell proliferation and activation of HCC progenitor cells and results in cancer promotion (95, 97). Like macrophages themselves, these cytokines play dual roles in liver cancer development by 1) promoting proliferation of cancer cells and 2) exacerbating liver injury to produce a protumor microenvironment.
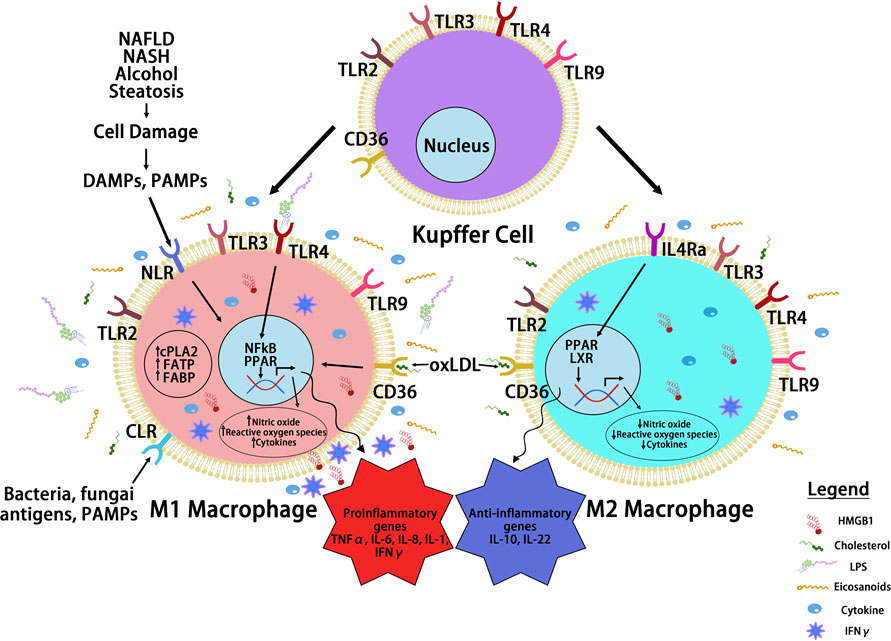
FIGURE 3
Figure 3 Macrophage Reprogramming in Steatosis Driven HCC. During liver inflammation, Kupffer cells and macrophages express scavenger receptors (SR) and pattern recognition receptors (PRR) to respond to pathogens and liver damages. Activation of PRR receptors by pathogen activated molecular pattern (PAMP) and damage activated molecular pattern (DAMP) molecules reprograms hepatic macrophages to produce inflammatory cytokines/chemokines. The binding of PRRs and SRs to steatotic induced PAMP and DAMPs reprograms hepatocyte macrophages. The reprogramed M1 macrophages produce a proinflammatory cytokines where the reprogrammed M2 macrophages produce anti-inflammatory cytokines to mediate the progression of steatosis to cancer. Toll like receptor (TLRs) and NOD-like receptors (NLRs) are two common PRRs used by PAMP and DAMP to induce macrophage reprogramming. Cluster of differentiation 36 (CD36) belongs to SR family of receptors and binds to oxidized LDL. Other PRR receptors include the C-type lectin receptors (CLRs) is also expressed on the reprogrammed macrophages. Binding of these receptors to their ligands such as lipopolysaccharide (LPS), high mobility group box 1 (HMGB1) and oxidized low density lipoprotein (oxLDL) activates the innate immune response and produce cytokines and chemokines that play important roles in tumorigenesis. It also activates nuclear factor kappa B (NF-κB) and proliferator-activated receptor (PPAR) and other liver nuclear receptors such as liver X receptor (LXR) regulates transcriptional reprograming of these macrophages.
Proinflammatory cytokinesSeveral proinflammatory cytokines appear to be induced during the development and progression from steatosis to liver injury to cancer (71, 75, 90, 100–106). In patients with chronic inflammatory and fibrotic liver diseases, analysis of classical CD14++CD16- monocytes in the liver found that they express both macrophage and dendritic cell markers with a high capacity for phagocytosis, antigen presentation, and regulatory T cell proliferation (103). They also secrete proinflammatory cytokines including TNFα, IL-6, IL-8 as well as IL-1 consistent with a role in the wound healing response where proinflammatory cytokines induce hepatocyte proliferation for tissue repair. In mice fed a Western diet, tumor progression is associated with a predominant M1 proinflammatory cytokine vs. the M2 pattern (83). In ALD, severe liver damage is also accompanied by significantly elevated M1 proinflammatory macrophage marker expression in C57Bl/6 mice, whereas less damage is observed in Balb/c mice where no change of M1 markers is found (104). In morbidly obese patients with NAFLD, reduced liver M2 anti-inflammatory macrophage marker expression (increased M1/M2 ratio) is associated with more severe steatosis. This reduced M2 macrophage phenotype also correlated with increased hepatocyte cell death and elevated serum levels of alanine aminotransferase (ALT), a clinical index of liver injury (107). Together, the proinflammatory cytokines secreted by macrophages in steatotic liver establishes a pro-inflammatory tissue microenvironment that can promote further liver damage and sustained inflammation.
One of the proinflammatory cytokines produced with steatosis, TNFα, plays a key role in liver carcinogenesis (16, 108). TNFα is a pleiotropic cytokine produced by many cell types with monocyte lineage cells being the primary source. In the liver, both Kupffer cells and infiltrating monocytes can produce TNFα in response to stimulation. TNFα produced by macrophages was found to promote cancer cell sphere formation in vitro (95). In this study, TNFα enhances the self-renewal abilities of the cancer cells. Consistently, in MUP-uPA mice fed with HFD, development of NASH and HCC are dependent on macrophage secreted TNFα. Knocking down TNFα Receptor 1 (TNFR1) significantly reduced liver damage and tumor formation (109). NFκB signal is implicated in this TNFR1 mediated hepatocyte death as deletion of IKKβ or NEMO, two NFκB signal modulators resulted in spontaneous progression of TNFα mediated hepatitis to cancer (110). In a DEN induced tumor model, deletion, or inhibition of TNFα resulted in reduced tumor incidence accompanied by suppressed activation and proliferation of hepatic progenitors via the TNFR2-STAT3 pathway (111). Consistent with a role of TNFα in liver regeneration, hepatocyte growth is also inhibited, resulting in a shorter lifespan even though tumor burden was reduced. In CCA, this effect of TNFα signal in chronic liver injury was shown to be mediated by JNK signaling and involves mitochondrial reactive oxygen species (ROS) production (112).
It was determined that hepatic IL-6 expression is significantly increased in the livers of patients with NASH (113). IL-6 signals through two pathways on target cell: classical signaling involves IL-6 binding to its receptor IL-6R on target cells. In the absence of IL-6R, IL-6 trans-signaling is induced, which involves an IL-6 binding to cleaved and soluble IL-6R provided by surrounding cells (114). During hepatocellular carcinogenesis, IL-6 trans-signaling pathway, rather than the IL-6 classic signaling contributes to the development of tumors by enhancing tumor proliferation through STAT3 and β-catenin activation and stimulating endothelial cell proliferation to promote tumor angiogenesis (115). Furthermore, IL-6 induces pre-cancerous progenitor cell proliferation and transformation into tumor initiating cells (97). IL-6 treatment in vitro led to early S phase entry in H4IIE HCC cells as shown by the reduced G0/G1 phase after treatment (116). IL-6 also contributes to the drastically different HCC incidence in male vs female mice treated with DEN (117). Recruitment of tumor-associated macrophages by the Yes-associated protein YAP, an oncogene overexpressed in a subset of HCC patients, also involves IL-6 signaling (118). Similar to the role of TNFα, IL-6 signals through STAT3 protect from chronic liver injury. However, the role of IL-6 in liver injury and tumorigenesis is also context dependent as IL-6 also protects from liver injury by promoting hepatocyte regeneration. In the multidrug-resistant gene 2 knockout (Mdr2-/-) mice where 50% of the mice develop tumors after chronic injury, IL-6 signal deficiency led to more severe steatosis and inflammation presumably due to the inability of hepatocyte regeneration/increased hepatocyte apoptosis after injury (101). Regardless, the resulting infiltration of macrophages promoted tumor growth and led to increased tumor burden (101, 119).
Other proinflammatory cytokines including IL-1, IL-8, IL-17, IL-18 and IFNγ may also be produced by macrophages to play similar roles in liver regeneration and sustain tumor cell growth. Like IL-6 and TNFα, IL-1 is commonly induced in steatotic livers when macrophage proliferation and infiltration are induced (17, 77, 84, 120) and is necessary for the whole spectrum of pathologies associated with steatosis, injury and cancer (104, 106). The expression of C-X-C receptor 2 (CXCR2), a receptor for IL-8, is upregulated in both HCC and iCCA. Targeting inhibition of CXCR2 results in reduced proliferation in Huh7 and HepG2 cells (121). The induction of liver IL-8 also provides signals for breast cancer cells to escape dormancy when they metastasize to the liver, suggesting that IL-8 indeed establishes a protumor environment in the liver (122). Blockade of IL-17 was shown to protect from liver injury including injuries induced due to steatosis (123). While macrophages may or may not be the primary source of IL-17 (124, 125), IL-17 does induce hepatic macrophage production of IL-6 and TNFα (126, 127). IL-18 is produced by THP-1 macrophages together with IL-1 in cultures exposed to hepatitis C virus (128). In the liver, administration of recombinant IL-18 induces severe liver injury concurrent with induced IFNγ secretion from NK cells (129). Delivery of neutralizing antibody targeting IL-18 reduced serum ALT levels and liver inflammation. Together, the proinflammatory cytokines produced by Kupffer cells and infiltrating monocyte derived macrophages establishes a sustained inflammatory environment to promote the growth of hepatocytes. This proinflammatory environment also acts on tumor initiating cells to propagate the genotoxic events, leading to tumor development.
Anti-inflammatory cytokinesMacrophage polarization was defined by IL-1β/iNOS producing macrophages as M1 and Arg-1/IL-10 expressing macrophages as M2 phenotypes. As the defining M2 cytokine, IL-10 is one of the best documented anti-inflammatory cytokines. In HFD feeding or alcohol induced liver injury, IL-10 is also induced (84, 108). It was proposed that the Kupffer cell production of IL-10 is also pro-regeneration and pro-survival for the hepatocytes (130). During the initial stage of chronic liver damage, liver macrophages also express C-X-C Ligand16 (CXCL16) to recruit NKT cells (131). This results in the formation of NKT and Kupffer cell clusters during liver steatosis. Clustered NKT-Kupffer cells secrete IFNγ and IL-10 (23). Another IL-10 family of cytokine, IL-22 also plays a role in NASH driven hepatocarcinogenesis. IL-22 levels gradually increase 5 months after the start of DEN treatment. It was concluded that continuous activation of STAT3 and CyclinD1 sustained IL-22 promoted cell proliferation (132). More recently, metformin, the antidiabetic drug was found to promote cell apoptosis through activation of Hippo signaling and to inhibit IL-22 induced tumor cell proliferation and invasion (133).
ChemokinesChemokines are released by Kupffer cells, liver sinusoidal endothelial cells and hepatic stellate cells to recruit infiltrating immune cells (134). Chemokine levels and their receptors are elevated in tissue and blood samples from patients with NASH and HCC compared with healthy and non-tumor controls (135–142). Among the two primary groups of chemokines, CC chemokines (CCLs) are known for their ability to recruit monocytes and lymphocytes, while CXC chemokines (CXCLs) are potent neutrophil attractants and can promote angiogenesis (143).
Upon ligand binding, Kupffer cells release CCL2 to recruit monocytes (144). In Ccl2 deletion mice, reduced inflammatory cell infiltration is observed (76). Inhibition of CCL2 with an RNA oligonucleotide that binds to CCL2 or neutralizing antibody for CCL2 led to reduced monocyte chemotaxis and reduced macrophage infiltration into the liver (82). These treatments resulted in reduced production of TNFα and IL-6, two macrophage produced cytokines. In NAFLD and NASH livers, macrophages also upregulate CCL3 and this induction of CCL3 facilitates macrophage infiltration and production of proinflammatory cytokines (135).
Kupffer cells also release CXCL1,CXCL2 and CXCL8 to recruit neutrophils (144). In HFD+Alcohol induced liver steatohepatitis, blockage of CXCL1 was found to reduce hepatic neutrophil infiltration and significantly inhibit liver injury (145). CXCL2 induction was shown to play a pivotal role in the recruitment of neutrophils in ConA induced hepatitis (146). In cholestatic patients, upregulation of CXCL8 and its receptors CXCR1/2 is associated with neutrophil infiltration whereas macrophage infiltration is associated with CXCL8 signal upregulation in non-cholestatic patients (147). This upregulation of CXCL8 signal plays important roles in the tumor microenvironment (122, 142). Furthermore, the macrophage derived CXCL9 and 10 are required for immune checkpoint therapy to block the infiltration of CD8+ T cells (148). The release of CXCL10 from macrophages is induced by steatosis (149) and deficiency of macrophage lipid receptor CD36 led to reduced release of CXCL10 in the liver (150). The role of CXCL5 in steatosis and liver cancer has drawn attention recently (151). Hepatic CXCL5 expression was higher in patients with severe fibrosis and cirrhosis (141). Multivariate Cox analysis of TCGA data identified that among 110 differentially expressed genes that were associated with HCC overall survival, CXCL5 and IL18RAP were the only 2 genes that predict the prognosis independently (142).
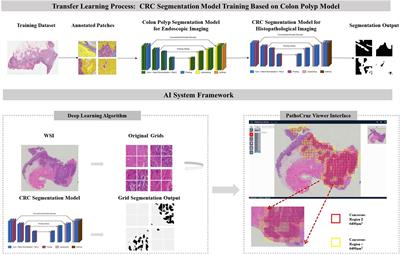
Macrophage reprogramming in steatosis driven HCCThe crosstalk of macrophage with hepatocytes is crucial for sustaining inflammatory signals during liver injury. In normal livers, macrophages contribute to normal hepatocytes function by regulating glucocorticoid signals (152). During liver inflammation, Kupffer cells and infiltrating macrophages express scavenger (SR) and pattern recognition receptors (PRR) to readily respond to pathogens and liver damage (153). Activation of PRR receptors by pathogen activated molecular pattern (PAMP) and damage activated molecular pattern (DAMP) molecules produced primarily by hepatocytes reprogram hepatic macrophages to produce inflammatory cytokines/chemokines that reverse the immune-suppressive liver environment and facilitate tissue repair (154). Scavenger receptors (SRs) are defined as macrophage receptors for modified lipids in foam cell formation but can also bind to other bioactive ligands (155). While binding of PRRs to ligands induces the release of pro- and anti-inflammatory cytokines and chemokines, uptake of modified lipids via SRs also leads to removal of the pathogen/damaged cells that present the recognized molecular patterns in addition to releasing inflammatory mediators (Figure 4).
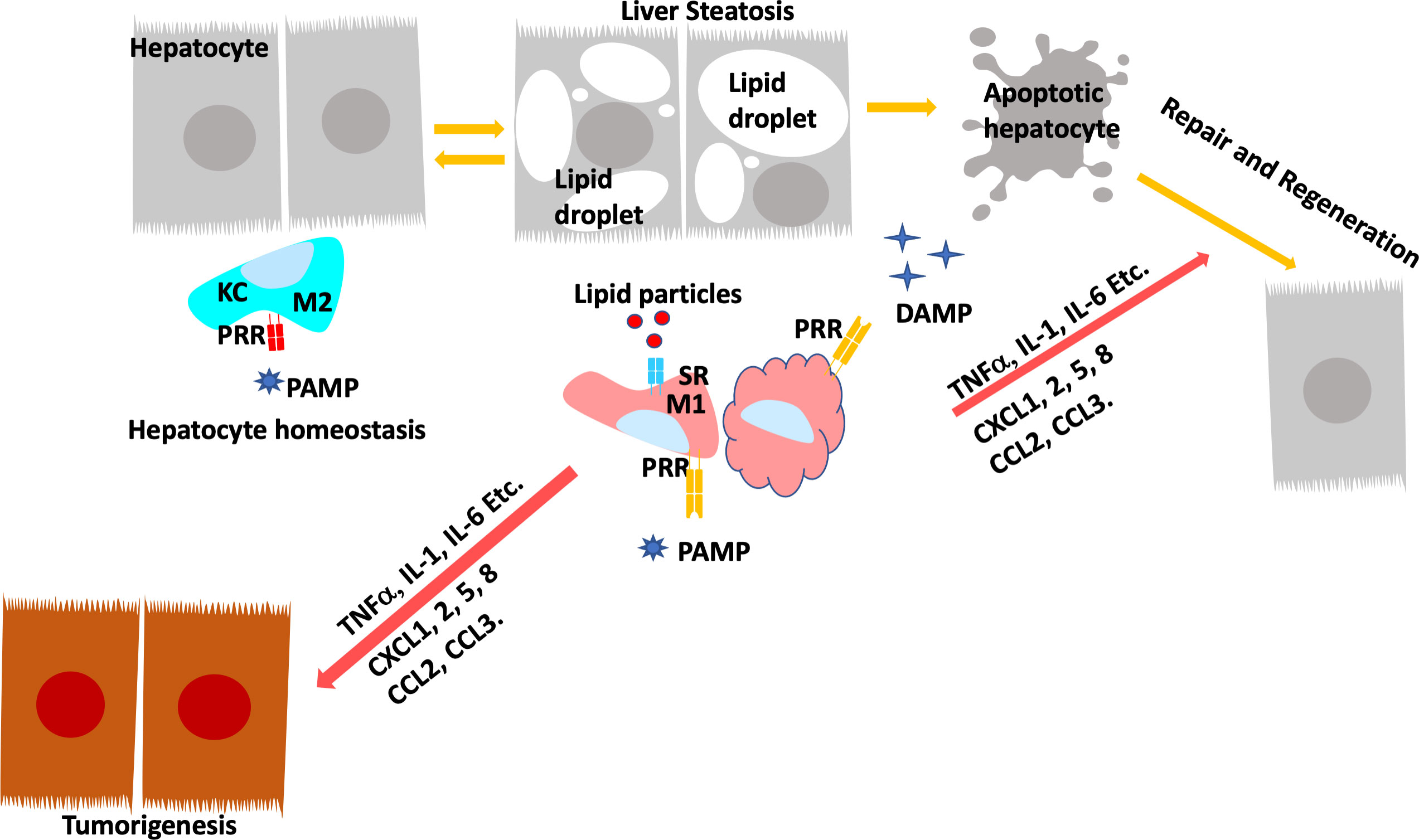
FIGURE 4
Figure 4 Programing of Hepatic Macrophages by PAMP and DAMP via PRR and SR. In normal, Kupffer cells recognize pathogen induced molecular patterns (PAMP) such as LPS coming through the portal vein. Kupffer cells clears these bacterial toxins without inducing inflammation to maintain hepatocyte homeostasis. During steatosis and steatotic injury, PRRs also bind to damage induced molecular patterns (DAMP) released by hepatocytes. The chronic injury induces proinflammatory responses from Kupffer cells as well as infiltrating macrophages. In addition, particles released by steatotic hepatocytes are also taken up by macrophages via scavenger receptors (SR). The binding of DAMP and lipid particles to PRR and SR induces the release of proinflammatory cytokines and chemokines including TNFα, IL-1, IL-6, CCL2 and 3 as well as CXCL1,2,5, and 8. These inflammatory mediators signals tissue repair and also promotes genotoxic events in liver cancer.
Pattern recognition receptorsMacrophages possess a number of different receptors that recognize intracellular and extracellular PAMPs and DAMPs as well as membrane bound ligands. This includes the TLR family of membrane receptors that play key roles in both innate and adaptive immune response. A cytosolic nucleotide-binding domain and leucine-rich repeat containing receptors (NOD-like receptors, NLRs) is another super family of PRR that is responsible for inflammasome activation which is essential for a successful immune response. The C-type receptors (CLRs) at the cell membrane recognize foreign antigens including bacterial and fungal antigens. Other PRRs including the 5’-triphosphate-RNA and dsRNA RIG-I-like receptors, as well as several DNA cytosolic sensors are also expressed in the liver microenvironment.
In NAFLD and ALD, steatosis induces chronic injury and hepatocyte damage. The damaged hepatocytes are the major source for DAMPs in the steatotic liver. For example, bile acid accumulation in hepatocytes triggers the assembly of NLR protein 3 inflammasome and the subsequent release of IL-1β that can bind to IL-1 receptors on macrophages. The TLR family members are high-affinity transmembrane receptors expressed on macrophages including Kupffer cells (156). The engagement of TLR4 with LPS triggers the sequential release of proinflammatory cytokines including TNF, IL-1, and IFN-β and other proinflammatory mediators such as the high mobility group box 1 (HMGB1) (157). During fatty liver diseases, free fatty acids also induce HMGB1 overexpression and secretion from hepatocytes. HMGB1 binds and activates TLR4 receptors on Kupffer cells and induce the release of proinflammatory cytokines such as TNFα and IL-6 (158). Similarly, during HFD feeding, hepatocytes release mitochondrial DNAs which stimulate Kupffer cell TLR9 receptors and subsequent TNFα secretion. Cholesterol laden lipid droplets formed within hepatocytes can also activate Kupffer cells through direct contact, this promotes IL-1β secretion in these Kupffer cells (159).
Scavenger receptorsThe distinct characteristic of steatotic liver injury is lipotoxicity. The accumulation of lipids in hepatocytes results in metabolic and oxidative stress that not only results in hepatocyte apoptosis but also directly signals inflammatory responses via macrophage cell surface receptors (55, 160, 161). During the pathogenesis of atherosclerosis, plaque formation is induced by the foam cells formed when macrophages scavenge modified low-density lipoproteins (LDL) and deposit them into the endothelial linings of blood vessels. Brown and Goldstein identified SR that are responsible for uptake of the modified LDLs (155). The family of scavenger receptors now are diverse and bind other DAMPs and PAMPs as well. In addition to modified and unmodified LDLs, other lipids such as cholesterols and phospholipids, bacterial pathogens, oxidative particles, and apoptotic cells are all scavenged by macrophages via these scavenger receptors (155). In NASH induced by feeding of Western diet, deletion of macrophage scavenger receptor MSR or type B1 scavenger receptor CD-36 led to reduced inflammation likely due to their effects on intracellular cholesterol trafficking in Kupffer cells (162, 163). In LDL receptor deficient (ldl4-/-) mice fed HFD, loss of CD36 or MSR resulted in reduced hepatic inflammation (162). In ConA induced liver injury, it was shown that CD36 sustains inflammation and expression of proinflammatory cytokines and is required for C-X-C ligand 10 induced apoptosis of hepatocytes (150).
Uptake of cholesterol via these SRs reprograms liver X receptor (LXR) regulated transcription in macrophages and attenuates the expression of anti-inflammatory genes (164, 165). In addition, the expression of macrophages CD36 and SR-B2 are also subjected to the transcriptional regulation by the orphan nuclear receptor peroxisomal proliferator-activated receptor (PPAR) (166–168). In THP-1 macrophages, it was shown that the downregulation of CD36 in macrophages likely resulted from reduced PPARγ regulated transcription when ratio of n-6/n-3 polyunsaturated fatty acids (PUFAs) is reduced (169). PPARγ has long been recognized as a potential receptor for PUFA produced eicosanoids (170). These effects of PUFAs on CD36 expression and the function of macrophages to produce inflammatory metabolites are at least partially mediated through the activation of PPARs by the bioactive eicosanoids produced from PUFAs (171–173). In fact, cyclooxygenase 2 (COX-2), one of the enzymes metabolizing PUFAs to eicosanoids is only expressed in tissue and infiltrating macrophages in the healthy liver (174). Activation of PPARγ by eicosanoids was found to sustain the production of TNFα, IL-1 and IL-6 induced by LPS and induce IL-10 downregulation in macrophages (175). In HFD induced NAFLD, loss of CD47, an inhibitor for macrophage activation and phagocytosis, leads to increased production of proinflammatory cytokines involving activation of PPARα (176). In Kupffer cells, LPS treatment induced TNFα and IL-6 is attenuated by PPAR agonist rosiglitazone (177). Thus, via regulation of PPARs, macrophages scavenge lipid particles to produce both pro- and anti-inflammatory cytokines (160). These PUFA derivatives including prostanoids, leukotrienes, HETES, EETs and lipoxins have all been indicated to promote a protumor inflammatory environment (178). During hepatocarcinogenesis, inhibiting COX-2 and epoxide hydrolase led to reduced “cytokine and eicosanoid storm”, resulting in cancer prevention (179). The treatment with lipoxin A4, a pro-resolving eicosanoid in inflammation, led to reduced HCC proliferation induced by activated macrophages (180). Together, macrophage engulfment of lipids via the scavenger receptors will result in increased production of PUFA derived eicosanoids. These eicosanoids can be inflammatory mediators on their own and induce the production of inflammatory cytokines/chemokines via the transcriptional activities of nuclear receptors such as PPAR and others. By producing the proinflammatory eicosanoids and cytokines, macrophages/Kupffer cells establish a pro-tumor microenvironment in the injured livers of NAFLD and NASH (181–183).
The therapeutic potential of targeting steatosis for liver cancer treatmentPathologically, 80% of liver cancer occurs in patients with underlying liver disease that displays lipid metabolic dysfunctions known as liver steatosis (184), a condition that develops in all obese individuals and is commonly associated with liver cancer (5). In a zebra fish model of HCC promoted by HFD, metformin the first line drug used for treatment in diabetes, reduced TNFα expressing pro-inflammatory macrophages leading to increase T-cell population in the livers, and inhibited cancer progression (185). In mouse HCC induced by DEN treatment, metformin treatment reduced the number of foci. This reduction was thought to be an effect of lowered hepatic expression of interleukin-22 and inhibition of YAP phosphorylation (133). The binding of metformin directly to the C-terminal of HMGB1 may also play roles in its anti-inflammatory and tumor suppressive functions (186). Statin, a cholesterol lowering drug has been proposed as treatment for chronic liver disease (187). NAFLD patients who take more than 600 cumulative daily doses of statin had a 70% reduction in hazards of developing HCC (HR, 0.30; 0.20-0.43) (188). Longer usage of more than 5 years and higher doses reduced the rate of NASH related HCC by 24-35% (189, 190). Both Metformin and Statin may target AMPK for their lipid reduction function (191). In a HFD model treated with DEN, AMPK activator reduced tumorigenesis and IL-6 signaling in the liver (192). Activation of AMPK also suppresses HCC progression and metastasis induced due to deficiency of FATP5 (fatty acid transporter protein 5) (193). Loss of the upstream kinase, LKB1 that phosphorylates and activates AMPK was also found to synergize with Pten loss to promote liver cancer development (194). Indeed, Sorafenib, the first line targeted therapy for HCC suppresses NASH through mechanisms involving alteration of mitochondrial uncoupling and subsequent activation of AMPK (195). These observations indicated that mitochondrial metabolism is an underexplored mechanism that may provide potential targets for HCC treatment as LKB-AMPK acts as primary cellular sensors of energy crisis to promote ATP production. Consistently, plasmas from NASH patients were found to contain high levels of mitochondrial DNA and these mitochondrial DNA signal through TLR9 to regulate hepatic inflammation, acting as a potential mechanism for how steatosis establishes the proinflammatory tumor microenvironment. In addition, targeting mitochondrial functions attenuates steatosis and inflammation in the liver (196, 197). Together, this evidence suggests that targeting steatosis via reducing lipid burden and/or altering mitochondrial function can impact liver cancer development.
The majority of liver cancer patients are diagnosed in the advanced stages of the disease, eliminating surgery or transplantation the only curative treatment for liver cancer. In patients with advanced disease, the combination of immune checkpoint (CPI) therapy such as anti PD-L1 antibody atezolizumab and the VEGF antibody bevacizumab has become the new standard of care. PD-L1 is highly expressed by liver macrophages in the tumor stroma (198). These macrophages repress the tumor-specific CD8 T-cell activity and induce their apoptosis through the Fas receptors to promote tumor growth (199, 200). Furthermore, Kupffer cells also stimulate the proliferation of antigen specific CD4+ Tregs and their release of IL-10 to inhibit the activities of cytotoxic T lymphocyte (91, 92). Additionally, prostaglandins produced by Kupffer cells may inhibit T cell activation (201–203). Together, activation of hepatic macrophages and their expression of PD-L1 appears to promote tumor escape by inducing an immune tolerance and reduce immune surveillance.
Patients with NASH and ASH respond poorly (median survival 5.4 months) to CPIs compared to those without steatosis (Median survival 11 months) (204). Given that CPI blocks the ability of macrophage/Kupffer cells to induce immunosuppressive environment in the liver, identifying the hepatic macrophage produced factors that allow the liver to escape this immune surveillance may be a key to future therapeutic development targeted at inflammatory tumor microenvironment associated with steatosis. As DAMP and PAMP that are present in the NAFLD/NASH livers, PRR and SR signals that controls the macrophage response to DAMP and PAMP are considered potential targets of intervention. A promising dietary intervention is n-3 fatty acids. Treatment with n-3 fatty acids was shown to inhibit both protein and mRNA levels of CD36 whereas n-6 fatty acids activate both (205, 206). In fat-1 transgenic mice fed STZ/HFD to induce NASH, ubiquitous expression of n-3 desaturase converts n-6 PUFAs to n-3 PUFAs and led to downregulation of CD36 and reduced liver damage (207). These dietary intervention studies suggest that targeting PRR and SR may be promising to reduce the tumor microenvironment and may work together with CPI to attenuate tumor growth in the liver.
One interesting discovery in CPI resistance is the role of the Wnt/β-catenin signal. The Wnt/β-catenin signaling pathway plays versatile roles in liver metabolism and tumorigenesis (32, 48, 208) due to its varied functions in different cell types in the liver. As such, upregulation of β-catenin allows tumors to escape CPI therapy and is one of the signals highly associated with CPI resistance together with steatosis (87). Interestingly, steatosis was found to induce macrophage expression of Wnt and the Wnt/β-catenin signaling mediates tumorigenesis in mouse models (32, 88). Thus, the induction of Wnt in macrophages by steatosis may play a role in the immune escape of these tumors. Further studies to elucidate how steatosis induces Wnt upregulation in macrophages is necessary to understand the resistance of steatosis associated liver cancer to CPI treatment.
Overall, liver cancer is the 6th most common type of cancer and the second leading cause of cancer deaths in the world with a median 10-year survival of just 11 months (209–211). In the liver, cancer development is highly associated with the development of steatosis and inflammation. Innate immune system and particularly Kupffer cells, the residence macrophages, act as the first responders following steatotic liver injuries. As such, targeting steatosis that show promising results in attenuating liver inflammation holds great potential in further therapeutic development as treatment of liver cancer (Table 1). Additionally, steatosis hinders CPI responses partially due to their effects on macrophages and macrophage production of inflammatory signals. Understanding how Kupffer cell are reprogramed to interact with innate immune system during the progression of steatosis is crucial for future therapeutic development targeted at overcoming resistance to current liver cancer therapy. Finally, identifying signals within tumor cells that respond to these protumor inflammatory signals produced by macrophages will result in novel therapeutic target that can overcome resistance to immunotherapy. In summary, targeting macrophages and macrophage interaction with tumor cells will provide therapeutic potential for steatosis-driven liver cancer treatment.
TABLE 1
Table 1 Current Therapy for HCC Treatment and Effects of Potential Lipid Modifying Therapy.
Author contributionsTT organized the writing of this manuscript. BLS edited the final content of the manuscript. All other authors contributed to either the writing or the art work of this manuscript and are listed alphabetically.
FundingBLS acknowledged funding from NIDDK DK131492 and NILM RML013315.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. New Engl J Med (2003) 348(17):1625–38. doi: 10.1056/NEJMoa021423
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Schutte K, Bornschein J, Malfertheiner P. Hepatocellular carcinoma–epidemiological trends and risk factors. Digestive Dis (Basel Switzerland) (2009) 27(2):80–92. doi: 10.1159/000218339
CrossRef Full Text | Google Scholar
4. Bray GA. Overweight is risking fate. definition, classification, prevalence, and risks. Ann New York Acad Sci (1987) 499:14–28. doi: 10.1111/j.1749-6632.1987.tb36194.x
CrossRef Full Text | Google Scholar
7. Nassir F, Rector RS, Hammoud GM, Ibdah JA. Pathogenesis and prevention of hepatic steatosis. Gastroenterol Hepatol (N Y) (2015) 11(3):167–75. doi: 10.3109/00365521.2015.1030687
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Tannapfel A, Denk H, Dienes H-P, Langner C, Schirmacher P, Trauner M, et al. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Virchows Arch (2011) 458(5):511–23. doi: 10.1007/s00428-011-1066-1
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Schattenberg JM, Schuppan D. Nonalcoholic steatohepatitis: the therapeutic challenge of a global epidemic. Curr Opin Lipidol (2011) 22(6):479–88. doi: 10.1097/MOL.0b013e32834c7cfc
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Myers S, Neyroud-Caspar I, Spahr L, Gkouvatsos K, Fournier E, Giostra E, et al. NAFLD and MAFLD as emerging causes of HCC: A populational study. JHEP Rep (2021) 3(2):100231. doi: 10.1016/j.jhepr.2021.100231
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Kudo A, Tanaka S, Ban D, Matsumura S, Irie T, Ochiai T, et al. Alcohol consumption and recurrence of non-b or non-c hepatocellular carcinoma after hepatectomy: A propensity score analysis. J Gastroenterol (2014) 49(9):1352–61. doi: 10.1007/s00535-013-0899-6
留言 (0)