
記住我










Cell-mediated immunity is perhaps the best understood alloimmune pathway. It is predominantly driven by T-cells following the presentation of alloantigens by antigen-presenting cells (APCs) via major histocompatibility complex (MHC) molecules, also called human leukocyte antigen (HLA) [7, 8]. HLA genes are highly polymorphic and large interindividual differences in allelic variants are the major immunological barrier to transplantation [9]. Two main modes play a role in this allorecognition. In the direct pathway, allogeneic MHC is presented directly to recipient T-cells by donor APCs. In the indirect pathway, recipient APCs phagocytise and present alloantigens to recipient T-cells as MHC–peptide complexes [8, 10]. MHC classes I and II are, respectively, recognised by CD8+ and CD4+ T-cells [7]. Following allorecognition, T-cells require secondary costimulatory signals, resulting in proliferation and differentiation [11].
Besides cytotoxic CD8+ T-cells, immunological responses are regulated by CD4+ helper T-cells, whose subtypes have different characteristics, ranging from cytolytic activity, activation of innate and other adaptive immune cells, to propagating or dampening inflammation [12, 13]. T-helper 1 (Th1) cells are a key source of interleukin (IL)-2, IL-12, interferon (IFN)-γ and tumour necrosis factor (TNF)-α, which drive a cytotoxic immune response. They are highly effective in activating macrophages, but can also cause direct allograft damage through Fas/Fas ligand-mediated cytotoxicity [6, 12, 14]. Abundant evidence demonstrates the ability of Th1 cells to mediate acute rejection and CLAD [6, 12, 15, 16]. Th2 cells can produce a variety of cytokines (IL-4, -5, -6, -10, -13), some of which downregulate further cytokine production, while others promote humoral immunity [12, 13]. For example, a Th1/Th2 balance in favour of Th2 and IL-10 can reduce rejection rates, and on the other hand, Th2 cells can accelerate rejection by releasing proinflammatory and potent profibrotic mediators such as IL-6 and IL-13 [12, 17]. Next to cytokines, a complex network of chemokines and their receptors, which function to recruit and activate various leukocyte subsets, are involved in the inflammatory processes leading to the development of BOS or RAS (e.g., CCR2/CCL2, CXCR2/ligand, CXCR3/ligand, and CCL5/RANTES interactions) [6, 18, 19] (figure 1).
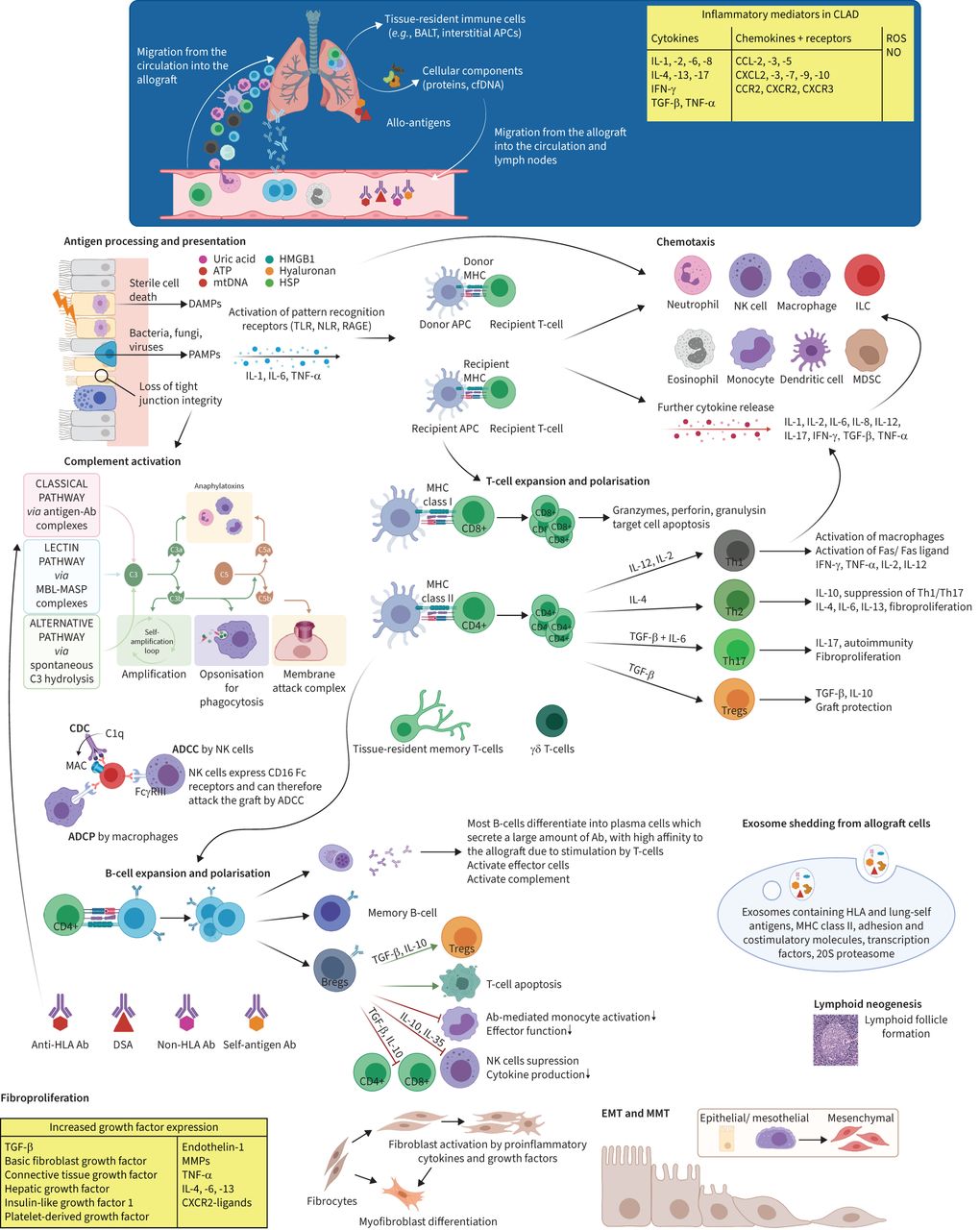 FIGURE 1
FIGURE 1 Key elements in the pathogenesis of chronic lung allograft dysfunction (CLAD). Overview of the pathogenesis of CLAD with some of the main immune mechanisms and cytokines involved. Tissue injury by alloimmune-dependent and -independent mechanisms induces the release of tissue damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs) and inflammatory cytokines, followed by antigen presentation to donor and host antigen-presenting cells (APCs) by pattern-recognition receptors. This is followed by an advanced interplay between innate and adaptive immune responses, with infiltration of innate and adaptive immune cells into the allograft. Activation of alloreactive T- and/or B-cells and suppression of regulatory T-cells further perpetuate an inflammatory milieu. Finally, fibrotic growth factors are upregulated and (myo)fibroblasts are activated, leading to deposition of extracellular matrix and, ultimately, fibrosis and allograft dysfunction. Ab: antibodies; ADCC: antibody-dependent cellular cytotoxicity; ADCP: antibody-dependent cellular phagocytosis; ATP: adenosine triphosphate; BALT: bronchus-associated lymphoid tissue; Breg: regulatory B-cell; CCL: C-C motif ligand; CCR: C-C motif receptor; CDC: complement-dependent cytotoxicity; cfDNA: cell-free DNA; CXCL: C-X-C motif ligand; CXCR: C-X-C motif receptor; DSA: donor-specific antibody; EMT: epithelial–mesenchymal transition; HLA: human leukocyte antigen; HMGB1: high-mobility group box 1; HSP: heat-shock protein; IFN: interferon; IL: interleukin; ILC: innate lymphoid cell; MAC: membrane attack complex; MASP: MBL-associated serine protease; MBL: mannan-binding lectin; MDSC: myeloid-derived suppressor cell; MHC: major histocompatibility complex; MMP: matrix metalloproteinase; MMT: mesothelial–mesenchymal transition; mtDNA: mitochondrial DNA; NK: natural killer; NLR: nucleotide-binding oligomerisation domain-like receptor; NO: nitric oxide; RAGE: receptor for advanced glycation end products; ROS: reactive oxygen species; TGF: transforming growth factor; Th: T-helper; TLR: Toll-like receptor; TNF: tumour necrosis factor; Treg: regulatory T-cell. Figure partially created with BioRender.com
Two other T-cell subtypes play important roles in the onset of CLAD. Firstly, Th17 cells, which secrete IL-6, IL-17, IL-22 and TNF-α, help to clear pathogens through recruitment and activation of neutrophils and macrophages, but are also associated with autoimmunity in cases of dysregulation or overproduction of IL-6 [13, 14]. Secondly, a unique subset of lymphocytes called regulatory T-cells (Tregs) have an important role in immune homeostasis [13]. Th17 and Tregs both develop from naive T-cells on stimulation by transforming growth factor (TGF)-β. IL-6, a proinflammatory cytokine, has a pivotal role in regulating the Th17/Treg balance, inducing the generation of IL-17-producing Th17 cells in concert with TGF-β, whilst inhibiting TGF-β-induced Treg differentiation [13, 14]. Both Th17/IL-17 and IL-6 are thought to be involved in the pathogenesis of CLAD, partly through endothelial cell activation and fibroblast activation and proliferation. IL-17 has also been shown to trigger a positive-feedback loop of IL-6 expression [13, 14, 20].
Tregs are essential components of the normal immune system and are responsible for maintaining homeostasis and balance activated immune responses. This is accomplished by the release of immunosuppressive cytokines (TGF-β, IL-10) as well as direct cell–cell interactions (e.g., regulation of dendritic cell maturation and function) [11]. These actions prevent excessive effector T-cell responses [14]. By promoting the differentiation and/or activity of IL-10-secreting T-cells, Tregs also protect against autoimmunity [21]. Tregs have been shown to reduce the onset of CLAD and to establish immune tolerance in animal models [11, 12, 22]. Increased proportions of Tregs, especially in the lung allograft, seemed to stabilise allograft function, while a decline of this cell population has been described in progressive CLAD [11, 23–27].
Apart from classical CD8+ cytotoxic cells, it has recently been shown that other cytotoxic cells with a senescent pattern are thought to be involved in CLAD development and associated with uncontrolled regulation by Tregs or immune checkpoints. For example, increased senescent T- and natural killer T-like lymphocytes with loss of CD28 expression were identified in BOS patients and correlated with increased expression of granzyme B, IFN-γ and TNF-α [28]. Brugière et al. [29] investigated whether the immune checkpoint HLA-G/immunoglobulin-like transcript (ILT)2 expressed by peripheral T-cell subpopulations could predict CLAD and found that an early increase after lung transplantation of cytotoxic CD4+CD57+ILT2+ T-cells, selectively inhibited by HLA-G, may be associated with CLAD onset. The importance of the role of these cells remains to be confirmed in large cohorts, but could open new avenues for targeted therapies.
Little is known about the precise role of other T-cell subsets including T follicular helper cells, Th9 cells and Th22 cells in the lung transplant setting yet, and the exact role of memory T-cells and γδ T-cells in the onset of CLAD remains also unclear [22, 30]. Memory T-lymphocytes are commonly viewed as an important barrier to long-term survival of organ allografts; however, Krupnick et al. [31] demonstrated an unsuspected role in lung allograft tolerance of central memory CD8+ T-cells, characterised by high surface expression of CD62 ligand and CD44, in a murine model. Further research on these T-cell subsets is warranted.
Humoral immunityTraditionally, CLAD was thought to be primarily a manifestation of T-cell-mediated immune responses; however, antibodies and pathological alloreactive B-cells play a significant role in CLAD [7, 32]. HLAs have a crucial role in immune surveillance by presenting peptides to T-cell receptors [8]. T-cells are required for the growth and maturation of antigen-specific B lymphocytes, which produce alloantibodies against mismatched MHC and minor histocompatibility antigens [11, 21]. The presence of donor-specific antibodies (DSAs) is strongly associated with CLAD, through alloimmune responses, complement activation, and complement-independent mechanisms [10]. Moreover, anti-HLA antibodies can induce the release of fibrotic growth factors, including platelet-derived growth factor, insulin-like growth factor-1 and TGF-β. These events culminate in the activation of myofibroblasts and extracellular matrix regeneration, thereby contributing to the development of CLAD [10, 11]. The onset of anti-HLA antibodies and subsequent complement activation are discussed in more detail in the section on antibody-mediated rejection (AMR).
Beyond their role in antibody production, B-cells can either contribute to or limit the development of CLAD via their regulation of T-cell immunity. B-cells influence T-cell responses through improved antigen presentation, co-stimulation, enhanced cytokine production, or induction of accommodation or tolerance [33–35]. However, the specifics of B-cell regulation in lung transplantation remain to be established, and the manifold and complex interactions between B- and T-cells are not yet fully understood [33]. A recent study demonstrated an increase in absolute peripheral B-cell count in patients with BOS, with a significant increase in specific subtypes of memory B-cells and a decrease in naive and transitional B-cells shown [36]. Similarly, Vandermeulen et al. [32] found higher levels of B-cells in BOS and RAS explant lungs and more lymphoid follicles in RAS tissue. The transformation of intragraft inflammatory infiltrates into tertiary lymphoid tissue, also called lymphoid neogenesis, probably also plays a role in lung allograft dysfunction, as has been reported in several other allograft types [10, 34].
Under some circumstances, humoral immune responses seem to cause little or no damage to the allograft. Accommodation describes a biological state in which the graft function remains stable despite alloantibodies or alloimmune responses, and is probably achieved by graft exposure to low concentrations of DSA or an altered affinity and/or specificity of the immune response [35]. Growing evidence demonstrates that B-cells also play a pivotal role in transplant tolerance [35]. Regulatory B-cells (Bregs) are thought to represent a stage of B-cell development before their differentiation into plasma cells and are potent inhibitors of the immune system, able to suppress allo- and autoimmune responses [33]. Bregs function, at least partly, through the production of IL-10, IL-35 and TGF-β, to suppress antigen presentation and cytokine secretion by APCs, T-cell proliferation, and actions from natural killer (NK) cells, neutrophils and other effector cells. Moreover, Bregs promote T-cell apoptosis and generation of Tregs by directly interacting with T-cell differentiation [35]. In addition to Bregs, other specific B-cell populations may be associated with long-term graft acceptance, such as IL-10 secreting CD9+ transitional B-cells as described by Brosseau et al. [37].
As a result, B-cells are increasingly acknowledged as crucial mediators at the centre of immune regulation with the power to enhance or inhibit allograft immunity.
High incidences of CLAD have been described in patients with previous episodes of AMR, and DSAs are a strong risk factor for acute cellular rejection (ACR), AMR and CLAD [38–42]. Numerous studies have attempted to identify DSA characteristics that correlate with worse outcome. Patients with anti-HLA antibodies prior to transplantation had increased risk post-transplant of developing antibodies to HLA and non-HLA molecules, AMR, CLAD and mortality, although some reports failed to substantiate this. Moreover, there is currently no consensus on the use of peri-operative desensitisation protocols in these patients [43–48]. Post-transplant de novo DSAs are also strongly linked to acute and chronic rejection and graft failure [40, 45–47, 49–54].
Detailed examination of DSA characteristics identified a greater risk for AMR, BOS and allograft loss in patients with DSAs against class II MHC molecules, especially DQ, compared to class I [40, 44, 55, 56]. A link between the number of total HLA mismatches and incidence of BOS has also been described [57]. Furthermore, the impact of circulating DSA depends on its ability to bind complement. Generally, complement-binding DSA was associated with worse CLAD-free and graft survival compared to non-complement-binding DSA [43, 49, 55]. Patients who cleared DSA after therapy had greater freedom from BOS and better survival rates than those who did not, which suggests that ongoing lung injury in the setting of persistent DSA results in accelerated graft dysfunction [46, 55, 58]. Based on these findings, many centres frequently monitor for DSA using highly sensitive immunoassays [10]. However, antibodies detected in the blood do not necessarily represent antibodies acting on the graft [34]. Importantly, it has been recognised that DSA might be absent in serum yet persist in allograft tissue [59].
Few studies have distinguished the effects of DSA or AMR on the development of BOS versus RAS. Until recently, AMR was believed to mainly occur early after transplantation as (hyper)acute rejection. However, AMR is increasingly seen beyond the first year post-transplant, which is likely partly due to increased awareness and implementation of sensitive detection methods. This raises the possibility of chronic AMR as cause for CLAD [60]. Moreover, patients with chronic AMR or persistent DSA seemed to be more prone to develop RAS than BOS [10, 46]. In patients with RAS, the level of tissue-bound DSA in the allograft seemed higher than in BOS, which might indicate a strong relationship with fibrosis [59]. It is appealing to consider whether RAS is an end-stage of chronic AMR, but definitive data are lacking to date and elevated B-cells, DSA and immunoglobulin G (IgG) were also seen in BOS, implying that chronic (less severe) AMR might also be a driving factor for CLAD phenotype BOS [32, 61].
AutoimmunityA critical feature of the immune system is to establish effective cell-mediated and humoral responses to foreign antigens while remaining unresponsive to self-antigens. This is checked centrally by negative selection of immature CD4+ T-cells recognising self-antigen and peripherally by anergy, apoptosis, and/or production of Tregs [11]. Mounting evidence has emerged that alloimmunity is not only directed against HLA, but also non-HLA and lung-associated self-antigens, suggesting a role for autoimmunity in the pathogenesis of CLAD [7, 21].
Collagen V (Col-V) and K-alpha 1 tubulin (Kα1T), two prominent self-antigens, are both components of small airways and are normally not exposed to the host immune system [7]. Col-V is found in the skin, lung epithelium and perivascular and peribronchial tissues, and placenta. It is an immunogenic self-protein that normally effectively masks its epitopes from the immune system because it is assembled in the same fibril as collagen I [11]. However, allograft injury (e.g., due to ischaemia-reperfusion injury, infection, DSA) enhances exposure of these antigenic proteins and results in the release of lung-derived autoantigens as soluble antigens, exosomes or apoptotic bodies. These are detected and then presented by APCs leading to the propagation of autoimmune responses through the Th17–IL-17 axis [11, 21]. This is possibly initiated by increased cleavage of Col-V due to upregulation of matrix metalloproteinases (MMP) 2 and 9 [21, 62], alongside loss of peripheral tolerance due to downregulation of Tregs and loss of IL-10 response to self-antigens [10, 62, 63].
Kα1T is a gap junction protein, essential for cytoskeletal structure and normal cellular function [11]. Similar to Col-V, repeated injury of the airway epithelium exposes Kα1T, resulting in expression of transcription and growth factors involved in fibroproliferation, suggesting that antibodies to Kα1T are directly pathogenic [21, 64].
A strong correlation between these antibodies and CLAD has been reported, in some instances in the absence of classic HLA antibodies. Conversely, autoantibody-mediated graft damage can trigger de novo DSA generation [64–66]. Although DSA can be transient, antibodies to self-antigens are often persistent. In patients with antibodies to both DSA and self-antigens, those who cleared DSA but had persistent autoantibodies were significantly more likely to develop BOS [67]. Moreover, patients with pre-existing autoantibodies had increased risk of developing de novo antibodies to DSA and non-HLA, AMR, primary graft dysfunction and CLAD [21, 65, 66]. Large cohort studies revealed that up to 30% of patients undergoing lung transplantation had pre-existing antibodies to lung self-antigens, primarily in patients with idiopathic pulmonary fibrosis and cystic fibrosis [65].
Taken together, both pre-existing and de novo lung self-antigens contribute to acute and chronic lung rejection through an interplay between allo- and autoimmunity, in which allograft immune responses may trigger autoimmune responses, which in turn further activate alloimmune responses. While alloimmunity may have initiated allograft injury, autoimmunity may ultimately contribute to the progression of CLAD [10, 21].
Several other autoantibodies have been described in other solid organ transplant recipients, and data on these autoantibodies are gradually becoming available in lung transplant recipients [68]. Firstly, antibodies to MHC class I-related chain A, expressed on endothelial cells and monocytes, have been associated with increased graft failure after kidney transplantation [68]. Likewise, Lyu et al. [69] and Angaswamy et al. [70] described a correlation between these antibodies and BOS. Secondly, the presence of angiotensin type 1 receptor or endothelin type A receptor antibodies correlated with allograft rejection in kidney and heart transplants [71]. Reinsmoen et al. [71] investigated the impact of these antibodies on graft outcome in lung transplantation and reported a trend toward higher ACR rates and an increased risk of de novo DSA. Follow-up time was not sufficient to observe CLAD outcome.
Innate immunityIt has been increasingly recognised that an advanced interplay between innate and adaptive immunity drives graft injury. Several innate immune pathways facilitate recruitment of inflammatory cells into the allograft and are key elements in the pathogenesis of primary graft dysfunction, acute rejection, and CLAD [72]. Innate immunity encompasses a broad spectrum of immune responses mediated by elements that are not reliant on gene rearrangement, including polymorphonuclear leukocytes, macrophages, NK cells and the complement system [72]. Innate recognition depends on pathogen- and damage-associated molecular patterns (PAMPs/DAMPs), recognised by pattern recognition receptors such as Toll-like receptors (TLRs), the receptor for advanced glycosylation endproducts and nucleotide-binding oligomerisation domain-like receptors [6]. DAMPs are endogenous molecules released from injured cells, such as high-mobility group box 1, heat-shock protein, hyaluronan, adenosine triphosphate, donor-derived cell-free DNA and mitochondrial DNA [72]. Recognition leads to immediate (sterile) inflammation, characterised by recruitment of mainly neutrophils and macrophages, upregulation of MHC expression and antigen presentation, followed by activation of the adaptive immune system (figure 1) [10]. The exact immune mechanisms in RAS have yet to be elucidated, but DAMPs appeared to be upregulated to a greater extent compared to BOS [73].
TLRs are transmembrane receptors mainly expressed by macrophages and dendritic cells, serving as a bridge between innate and adaptive immunity because of their ability to induce T-cell responses [10]. On the other hand, TLRs might contribute to CLAD directly [10]. For example, TLR4 signalling can induce fibroblast activation together with TGF-β, and in the case of sustained innate immune activation, the process of fibroblast activation might persist, leading to excess repair and fibrotic tissue remodelling [72].
Neutrophils play an important role not only in innate immunity, but also by enhancing antigen presentation and Th1-driven alloimmune responses [7]. Elevated bronchoalveolar lavage (BAL) and allograft neutrophilia have been repeatedly observed in patients with BOS and RAS, and early or persistent BAL neutrophilia correlated with subsequent CLAD occurrence [32, 74–79]. The relevance of neutrophils was further supported by the emergence of neutrophilic reversible allograft dysfunction, characterised by IL-17-mediated airway neutrophilia, in which azithromycin was able to attenuate pulmonary function decline [80]. IL-17 can induce IL-8, a major neutrophil chemo-attractant. Multiple studies demonstrated higher levels of BAL IL-8 in BOS patients with a correlation between neutrophils and IL-8 levels [77, 79]. IL-8 is secreted by alveolar type II epithelial cells, bronchial epithelial cells and macrophages after the release of proinflammatory cytokines [81]. In some patients, neutrophilia was not suppressed or redeveloped despite azithromycin, suggesting a non-IL-17-dependent pathway. Indeed, Vandermeulen et al. [80] found worse CLAD-free and overall survival in those patients, possibly driven by increased levels of IL-1β and IL-1β-induced proinflammatory cyto-/chemokines (e.g., IL-6, IL-8, macrophage inflammatory proteins, eosinophil attractants). Suwara et al. [78] also demonstrated an increase in IL-1α and IL-1β in patients with persistent airway neutrophilia. Activated neutrophils have remarkable potential to cause tissue damage through a variety of mechanisms: 1) release of large quantities of reactive oxygen species, 2) release of cytokines, 3) activation of hydrolytic enzymes and proteases, 4) expression of MMP that leads to degradation of collagen matrix [82]. An additional mechanism of neutrophil-mediated injury is the formation of neutrophil extracellular traps (NETs), a process known as NETosis. NETs are extracellular networks of DNA clad with granular proteins that were cast out from neutrophils and are thought to be an effector function of neutrophils [82] (figure 2).
 FIGURE 2
FIGURE 2 Non-alloimmune factors contributing to chronic lung allograft dysfunction (CLAD). Simplified representation of pathways involved in non-alloimmune mechanisms which may contribute to CLAD onset. APC: antigen-presenting cell; ATP: adenosine triphosphate; CM: classical monocyte; Col-V: collagen V; DAMP: damage-associated molecular pattern; dd-cfDNA: donor-derived cell-free DNA; DSA: donor-specific antibody; GORD: gastro-oesophageal reflux disease; HMGB1: high-mobility group box 1; ICAM-1: intercellular adhesion molecule 1; IFN: interferon; IL: interleukin; IRI: ischaemia-reperfusion injury; Kα1T: K-alpha 1 tubulin; MMP: matrix metalloproteinase; mtDNA: mitochondrial DNA; NCM: non-classical monocyte; NET: neutrophil extracellular trap; PAMP: pathogen-associated molecular pattern; PECAM-1: platelet endothelial cell adhesion molecule 1; ROS: reactive oxygen species; TNF: tumour necrosis factor. Figure partially created with BioRender.com
Eosinophils have also been implicated in the pathological process of CLAD. Two decades ago, Scholma et al. [83] had already noted that BAL eosinophilia correlated with increased BOS risk (RAS was not yet identified then). Likewise, a more recent study demonstrated a significant correlation between BAL eosinophilia and the development of CLAD, in particular RAS, and mortality [84]. The same group also found higher eosinophil levels in allograft tissue from RAS patients, and worse CLAD-free survival in patients with high blood eosinophils [32, 85]. Additionally, Darley et al. [86] demonstrated that detection of eosinophils on transbronchial biopsies was independently associated with an increased risk of CLAD and mortality. The actions of eosinophils are thought to be secondary to profibrotic features, by attracting fibroblasts and stimulating TGF-β release, as well as through toxic effects on airway epithelial cells (e.g., increased membrane permeability, ciliary damage) [84, 86]. Conversely, translational data from animal models recently illustrated a role for eosinophils in the downregulation of alloimmunity, potentially by the release of suppressive molecules or interactions with dendritic cells and lymphocytes [87]. These immunosuppressive effects are presumably exerted by a different subtype of eosinophils, such as tissue-resident eosinophils, although this needs to be further elucidated [87].
NK cells act as the first line of defence against infected or transformed cells and can directly respond to alloantigens and non-self cells through an arsenal of effector functions that are vital in innate–adaptive bridging [88, 89]. Increased numbers of activated NK cells were found in the lungs of CLAD patients, with corresponding peripheral blood depletion, suggesting systemic activation and subsequent migration into the allograft tissue [89]. Once activated, NK cells release a wide range of cytolytic proteins, such as granzymes and perforin, and chemotactic cytokines such as IFN-γ and TNF-α, which were found to be upregulated in CLAD [90]. Through the release of these cytokines, NK cells commit T-cells, skew immune responses to Th1, increase MHC class I and II expression, and induce graft infiltration by macrophages, dendritic cells and neutrophils [90]. Moreover, NK cells’ upregulation of Fc-receptors plays an important role in antibody-dependent T-cell-mediated cytotoxicity [88].
There is mounting evidence that NK cells have crucial and sometimes opposing roles in lung allograft rejection, due to either activating or inhibitory actions through different NK receptors [88]. NK cells might enhance CLAD through the above-described cytotoxic and inflammatory effects. On the other hand, it has been postulated that they may promote graft tolerance through depletion of donor APC and alloreactive T-cells via killer immunoglobulin-like receptors or possibly via IL-15–IL-15Ra complex expansion [88, 91]. Nonetheless, the exact mechanisms by which NK cells contribute to CLAD remain to be investigated.
Other innate lymphoid cells (ILC1, ILC2, ILC3) are a recently recognised and understudied group of immune cells, which are difficult to analyse because of their tissue-resident and lineage negative features. However, they can exert different actions such as type 1 immunity with macrophage activation and cytotoxicity, type 2 immunity and the formation of tertiary lymphoid structures by their different subtypes via the release of IFN-γ, granzymes, perforin, TNF-α, IL-13, IL-17, etc. [92, 93]. As such, it does not seem unlikely that they contribute to the pathogenesis of CLAD, and further investigation is warranted [94–96].
The complement system is a complex immune surveillance system made up of a cascade of multiple proteins, crucial in innate defence, and it plays a role in adaptive immunity via cell-mediated and humoral processes [97]. There are three different activation pathways (classical, lectin and alternative), all leading to the formation of a membrane attack complex which induces cell lysis. Furthermore, it stimulates immune complex clearing by opsonisation and, during activation of the complement cascade, signalling components known as anaphylatoxins are released and are capable of summoning various other innate and adaptive immune cells by stimulation of proinflammatory cytokines and chemotaxis [97]. Complement activation plays a role in the pathogenesis of primary graft dysfunction and AMR, which are risk factors for CLAD, but a direct contribution in the pathogenesis of CLAD is also assumed. Deposition of complement factors C1q, C3d and C4d in lung allografts was found to be independently associated with CLAD [97]. Higher levels of complement and IgG deposition were found in RAS compared to BOS patients, pointing to the role of humoral immunity and activation of B-cells in RAS, and the possible overlap between AMR and RAS [98, 99]. Several studies yielded some evidence that mannose-binding lectin, part of the lectin pathway, is involved in CLAD development. Higher levels were found in BOS versus stable patients, and presence of mannose-binding lectin at 3 and 6 months post-transplant correlated with later onset of BOS [97].
In conclusion, innate immune responses provide an early, robust trigger that augment adaptive alloimmunity, ultimately promoting CLAD development.
ExosomesRecently, exosomes have begun to attract attention as a trigger in CLAD development through activation of cellular and humoral immunity. Exosomes are dual-layer membrane vesicles which can contain HLA and lung self-antigens, adhesion and costimulatory molecules, MHC class II molecules, transcription factors, and 20S-proteasome. They are shed from allograft cells after lung injury and are highly efficient in presenting antigens to the immune system [100–102]. Exosomes have been shown to induce T-cell-mediated immune responses, and the induction and continuous release of exosomes from the allograft may stimulate the process of CLAD [21]. Furthermore, a recent animal study demonstrated the ability of exosomes, derived from lung transplant recipients with respiratory viral infections, to induce epithelial-to-mesenchymal transition (EMT) [103]. The role of exosomes in promoting EMT has been highlighted in cancer research and could be another way by which exosomes might initiate the process leading to CLAD [103]. Several studies demonstrated higher levels of exosomes, which also contained more of the aforementioned factors, in BOS patients [101, 104]. Furthermore, Sharma et al. [102] found that increased levels of circulating exosomes preceded the onset of BOS and could be detected 6–12 months before diagnosis.
Genetic variants associated with CLADSeveral donor- and recipient-related genetic variants may contribute to the development of CLAD [4]. Specific single nucleotide polymorphisms in TLR2, -4 and -9, were associated with a higher incidence of BOS [105]. Other types of polymorphisms in TLR4 correlated with a reduced risk of acute rejection and a trend toward reduced onset of BOS [106]. These findings again reinforce the importance of the link between innate immune responses and alloimmune response in the development of CLAD.
A polymorphism in HLA-G seemed to have a protective role by modulating cytotoxic T-cells and NK cells, while a specific HLA-E allele negatively influenced CLAD onset [107]. There is some evidence that functional polymorphisms in the genes of CD14, dectin-1, IFN-γ, IL-6, IL-17A, killer immunoglobulin-like receptors, mannose-binding lectin, MMP-7 and TGF-β1 are linked to CLAD development [108–110]. Regarding donor-related polymorphisms, gene polymorphisms in surfactant proteins, donor Clara cell secretory proteins, mannose-binding lectin and CD59 correlated with increased CLAD risk [97, 111–114].
In general, these genetic variants affect the innate defence system, altering immune responses to injury, possibly increasing susceptibility for airway inflammation or allograft infection, thereby contributing to the pathogenesis of CLAD [4].
Alloimmune-dependent risk factorsAcute cellular rejection and lymphocytic bronchiolitisAlloimmune-dependent factors such as ACR and lymphocytic bronchiolitis (LB) are strongly linked to CLAD [6]. ACR has been studied in more detail, but independent of acute vascular rejection, the onset and severity of LB is also associated with long-term outcomes after lung transplantation and an increased risk of BOS and death [127]. ACR and LB are driven by T-lymphocytes and many actions are similar as described in CLAD such as a predominance of Th1 cells with increased production of IFN-γ, IL-2 and TNF-α, activation of macrophages, direct allograft damage through Fas/Fas ligand-mediated cytotoxicity, and reduced Tregs [16, 25, 122]. There is currently too little evidence, but donor tissue-resident memory T-cells may play a protective role in ACR [22].
Although T-lymphocytes are regarded as the main culprit in ACR, other mechanisms contribute as well. It is well known that increased leukocytes, including lymphocytes and neutrophils, are found in BAL and tissue of patients with ACR, and increasingly more awareness is given to eosinophils and NK cells [84, 128–131]. Not much is currently known about the role of B-cells in acute rejection, but a recent study reported a decrease in the number of Bregs in peripheral blood and BAL during acute rejection and the role of B-cells in local lymphoid follicle formation could also be of importance in LB [16, 132]. It is worth noting that HLA antibodies do not only appear to be involved in the onset of AMR and CLAD, but also in ACR and LB [133, 134]. On the other hand, ACR and LB may predispose to de novo DSA [135].
In LB, there is convincing evidence of an IL-17-mediated pathway, which triggers IL-8-driven neutrophilic airway inflammation [130, 136]. Verleden et al. [136] found that patients with LB had significantly more IL-17+ cells on transbronchial biopsies compared to patients with ACR, and the number of IL-17+ cells correlated with BAL neutrophilia. Not Th17 cells, but CD8+ T-cells were the major source of this IL-17 production, which could be attenuated by azithromycin [137].
Ultimately, the alloreactive T-cell response and IL-17-mediated inflammation generate a profibrotic environment which can contribute to CLAD [130].
Antibody-mediated rejectionAMR results from the recipient's immune system recognising pre-existing or de novo antibodies to HLA, non-HLA or self-antigens [60]. Pre-existing anti-HLA antibodies may arise after prior sensitizing events such as pregnancy, blood transfusion or organ transplantation [41]. Risk factors for de novo DSA are only beginning to be identified. It is postulated that immunising events (e.g., transfusion, ACR) and lung injury (e.g., ischaemia-reperfusion injury, allograft infection) upregulate the expression of HLA molecules, thereby increasing the graft's immunogenicity [60]. Antibodies may develop to MHC class I antigens (HLA-A, -B, -C) which are expressed on nearly all nucleated cells, or MHC class II antigens (HLA-DQ, -DR, -DP) on professional APCs [41].
The binding of antibodies to directly accessible allogenic targets expressed by endothelial cells activates the classical complement pathway. This begins with binding of C1q, and eventually leads to membrane attack complex formation and cytotoxicity [34, 41]. Deposition of complement and IgG in lung allograft tissue have both been demonstrated [41, 138]. Activation of endothelial cells leads to the release of adhesion molecules and cytokines that, together with anaphylatoxins C3a and C5a, attract neutrophils, monocytes and NK cells to the allograft, propagating inflammation and graft injury [34, 41]. Complement-mediated allograft injury is a defining pathophysiological characteristic of AMR. However, graft injury can also occur independently of complement pathways through antibody-dependent cell-mediated cytotoxicity [88]. The latter is likely mediated by NK cells, recognising antibodies through their Fc-receptor, CD16, although antibodies can also bind the Fc-receptor of myeloid cells such as macrophages and neutrophils. In this way, antibodies bridge the innate and adaptive arms of the immune system [88].
Both complement-dependent and -independent mechanisms lead to the production of IFN-γ and other proinflammatory cyto-/chemokines, increased MHC expression, recruitment of leukocytes and platelets, amplification of innate and adaptive immunity, and upregulation of adhesion molecules and fibroblast growth factor receptor on endothelial cells. All these mediators contribute to microangiopathy, tissue injury and graft dysfunction [9, 41, 88].
Not all patients with DSA develop AMR, the clinical relevance of DSA may depend on the variable pathogenicity of IgG subclasses. Complement-binding IgG (IgG1, IgG3) seemed more damaging than non-complement-binding IgG (IgG2, IgG4) [43, 55]. Higher rates of early BOS were found in cases of increased C3d and C4d deposition early after transplantation [139]. Similarly, DSA-positive patients with increased C3d deposition had lower graft survival than those without C3d activation [138].
Although AMR might be a reversible cause of acute graft dysfunction, it generally portends a poor prognosis with a high incidence of CLAD amongst survivors and worse long-term survival compared to ACR [60].
留言 (0)