
記住我
Mismatch repair (MMR) deficiency is associated with tumor development and high microsatellite instability (MSI-H), and these phenotypes are observed in more than 15 different tumor types and frequently present in colon, endometrial, and gastric cancer.1 Due to their inability to repair replication-associated errors, MSI-H/mismatch repair-deficient (MMRD) tumors harbor large numbers of single-nucleotide substitutions and frameshifts and are characterized by a high tumor mutational burden (TMB).2 3 As a consequence, these tumors often have increased expression of neoantigens, high infiltration with activated CD8+ cytotoxic T lymphocyte as well as activated Th1 cells, which underlie immunogenicity and clinical responsiveness to immune checkpoint blockade (ICB) therapy.4 Indeed, programmed death 1 (PD-1) blockade has emerged as an effective therapy for patients with MSI-H/MMRD metastatic colorectal and non-colorectal cancers that are refractory to standard chemotherapy combinations.5–7 Such clinical responses have contributed to the approval of the anti-PD-1 (αPD-1) monoclonal antibodies (ie, pembrolizumab and nivolumab) for pediatric and adult patients with unresectable or metastatic MSI-H/MMRD solid tumors regardless of the tumor histology.8 However, as recently reported from the phase II KEYNOTE-158 study, which enrolled 233 patients with 27 different tumor types with microsatellite instability (MSI), the responsiveness of MSI cancers to αPD-1 monotherapy is very variable, with a response rate ranging from 0% in brain tumors to 57.1% in endometrial cancers.9 This data suggests that intrinsic and/or extrinsic factors may hamper αPD-1 therapy response. However, the underlying mechanisms remain elusive.
One of the critical factors that can influence the efficacy of immunotherapy is the tumor microenvironment (TME).10 11 While many studies have reported typically high lymphocyte infiltration in primary MSI-H tumors,4 little is known about other immune cell populations that may limit the antitumor immune response. Blood cells, such as neutrophils, monocytes and platelets have been associated with inflammatory responses, and the neutrophil-to-lymphocyte ratio (NLR) and platelet-to-lymphocyte ratio (PLR) have been used as inflammatory markers to predict outcomes in various malignancies.12 13 Several studies have evaluated the role of inflammatory cell ratios (eg, NLR and PLR) as predictive biomarkers in patients with solid tumors treated with ICBs, and higher NLR and PLR ratios at baseline are associated with treatment failure.14 Whether these inflammatory markers are associated with ICB resistance in MSI-H/MMRD tumors remains unclear.
In this study, we addressed this question at the preclinical and clinical levels. We first generated different MMRD mouse tumor models, including one with abnormal neutrophil elevation, and longitudinally explored the impact of this immune cell population on response to αPD-1 treatment in hypermutated tumors. We then monitored the dynamic changes in the NLR ratio in a cohort of 104 patients with 18 different MMRD tumor types treated with anti-programmed death ligand 1 (αPD-L1) to determine whether changes in the NLR during treatment could predict resistance to αPD-1 therapy regardless of the type of MSI tumors.
Materials and methodsMouse cell linesMouse 4T1 and CT26 cell lines were purchased from the American Type Culture Collection (CRL-2539 and CRL-2638, respectively) and were cultured at 37°C under 5% CO2 in RPMI-640 medium supplemented with 10% fetal calf serum, 100 U.mL–1 penicillin (Gibco) and 100 mg.mL–1 streptomycin (Gibco). Cell lines were regularly tested for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (LT07-418, Lonza).
Msh2 knock-out clones generated by CRISPR-Cas9‘All in one’ CRISPR-Cas9 plasmid targeting Msh2 exon 3 was provided by TACGENE (MNHN, Paris) with the guide RNA sequence: 5’-GGTATGTGGATTCCACCCAGAGG-3’. Both CRISPR-Cas9 plasmid and peGFP-C3 plasmid (Clontech) were transfected at a 9:1 ratio in 4T1 cells using Cell Avalanche reagent (EZ Biosystems) according to the manufacturer’s instruction. For CT26 cell line, the same strategy of transfection was carried out with JETPEI reagent (Polyplus) according to the manufacturer’s instruction. After 48 hours, GFP+ cells were fluorescence-activated cell sorting (FACS) sorted at one cell per well in 96-well plates then single cells were expanded.
Cell lysis and western blotFor validation of Msh2 inactivation, CRISPR-edited clones were lysed in sodium dodecyl-sulfate (SDS) lysis buffer (50 mM Tris pH 7.5, 20 mM NaCl, 10 mM MgCl2, 0.1% SDS, anti-proteases cOmplete cocktail from Roche) supplemented with 20 U/mL benzonase (Millipore) for 10 min at room temperature. Proteins were quantified with Bradford assay and denatured in Laemmli buffer. Proteins were separated on 8% acrylamide SDS–polyacrylamide gels and transferred on PVDF membranes (Millipore). Membranes were blotted with antibodies directed to the following proteins: MSH2 (rabbit, #Ab70240, Abcam, 1/5000), β-actin (mouse, #A5441, Sigma Aldrich, 1/10,000).
Genetic validation of Msh2 knock-out clonesGenomic DNA was extracted from CRISPR-edited clones using NucleoSpin Tissue (Machinery Nagel) then PCR was performed using Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific) with the following primers: for 5’-AGACTGACAGGCATGATTTTGT-3’; rev 5’-GGAAATACAGGGGAAGGGGTAT-3’. Cloning of PCR products was processed with CloneJET PCR Cloning kit (Thermo Fisher Scientific) and verified by Sanger sequencing.
Generation of MMRDint and MMRDhi cells linesBoth 4T1 and CT26 MMRD derivatives were serially passaged in vitro three times a week. The culture conditions were the same as for parental cell lines (37°C under 5% CO2 in RPMI-640 medium supplemented with 10% fetal calf serum, 100 U.mL–1 penicillin (Gibco) and 100 mg.mL–1 streptomycin (Gibco)). The TMB as well as indels were monitored over time. MMRD cells passaged for 6 weeks are hereinafter referred to as MMRD-intermediate cells (MMRDint) and MMRD cells that were passaged for more than 15 weeks are referred to as MMRD-high cells (MMRDhi).
Mouse studiesFemale BALB/c mice aged of 6 weeks were purchased at ENVIGO and were maintained in the animal facility of Gustave Roussy. Experiments were performed in accordance with French government and institutional guidelines and regulations. Inoculation of 50,000 4T1 tumor cells were done in the mammary fat pad (left, n°4) of BALB/c mice. Inoculation of 500,000 CT26 tumor cells were done subcutaneously into the right flank of mice. Tumor were measured twice weekly and tumor volume and was calculated as follows: length×width2. Mice were euthanized when the tumor size was ≥1500 mm3 or boundary points were reached according to the French and European laws and regulations for the use of mice for scientific purposes.
Exome analysisGenomic DNA extraction from mouse tumors was performed using AllPrep DNA/RNA micro Handbook (Qiagen). The exomes were sequenced according to the manufacturer protocols (BGI Tech solutions, Hong Kong) with BGISEQ-500 sequencer. We mapped reads using BWA-MEM software to the mm9 mouse reference genome and then used the standard GATK best practice pipeline15 process the samples and call somatic mutations. PCR duplicates were removed and base quality scores recalibrated using MarkDuplicates and BaseRecalibrator tools (GATK package).16 Somatic SNVs and INDELs were called and filtered using GATK tools Mutect2, FilterMutectCalls and FilterByOrientationBias and functional effects of mutations were annotated with ANNOVAR.17 Quality control of FASTQ and mapping was done with FastQC, samtools,18 GATK HSMetrics and MultiQc.19 The processing steps were combined in a pipeline built with snakemake scripts.20
Somatic mutations with PASS flag from GATK Mutect2 were additionally filtered to be supported by at least one read from each strand and at least three reads in total. The contribution of the COSMIC mutational signatures was assessed using MutationalPatterns package.21 Classification of mutations and insertions/deletions by types was done with SigProfilerMatrixGenerator.22
To calculate TMB, the total number of somatic non-synonymous mutations was normalized to the total number of sequenced megabases.
Mice treatmentMice received three intraperitoneal (i.p) injections of αPD-1 (clone RMP1-14; 250 µg/mouse) and/or αCTLA (clone 9D9, 100 µg/mouse) or isotype controls when tumor volume reached 70–100 mm3. αLy6G (clone 1A8, 100 µg/mouse) depleting antibody or isotype control was injected i.p when tumor volume reached 50–70 mm3 every 3 days. αCD-25 treatment (PC-61.5.3, dose 200–300 µg/mouse) depleting antibody or isotype control, was injected i.p when tumor volume reached 50–70 mm3 every 3 days. Injections was carried out 1 day before ICB administration. Antibodies are described in Key Resources table 1.
Flow cytometry analysisFor mouse tumor dissociation, tumor specimens were cut into small pieces and digested in RPMI medium containing 25 µg/mL Liberase (Roche) and 150 IU/mL DNaseI (Roche), and subsequently subjected to gentle MACS dissociation (Miltenyi Biotech) for 30 min at 37°C. Cells were then filtered through a 100 µm cell strainer, Fc receptors were blocked for 15 min at 4°C using with αCD16/32 functional grade purified antibodies (eBioscience). For FoxP3 and KI67 staining, cells were fixed and permeabilized, after cell surface staining, according to the manufacturer protocol (eBioscience). Antibodies are described in Key Resources (table 1).
For longitudinal blood monitoring, blood was collected twice a week from the submandibular vein of mice using minivette POCT. For whole blood staining, red cells were lysed using a mix of VersaLyse lysing solution +Fixative solution (1000:25, Beckman Coulter) during 15 min at room temperature then immunostaining was performed after Fc receptor blocking. Cells were then washed two times with phosphate buffered saline. Blood and dissociated tumor samples were acquired on a Gallios Cytometer (Beckman Coulter) and analyzed using Kaluza software (Beckman Coulter). The myeloid-to-lymphocyte ratio was calculated by dividing myeloid cells (%) by lymphocytes (%). These populations were identified in live CD45+ cells according to surface structure as described in online supplemental figure S1. The gating strategy for neutrophils is described in online supplemental figure S1.
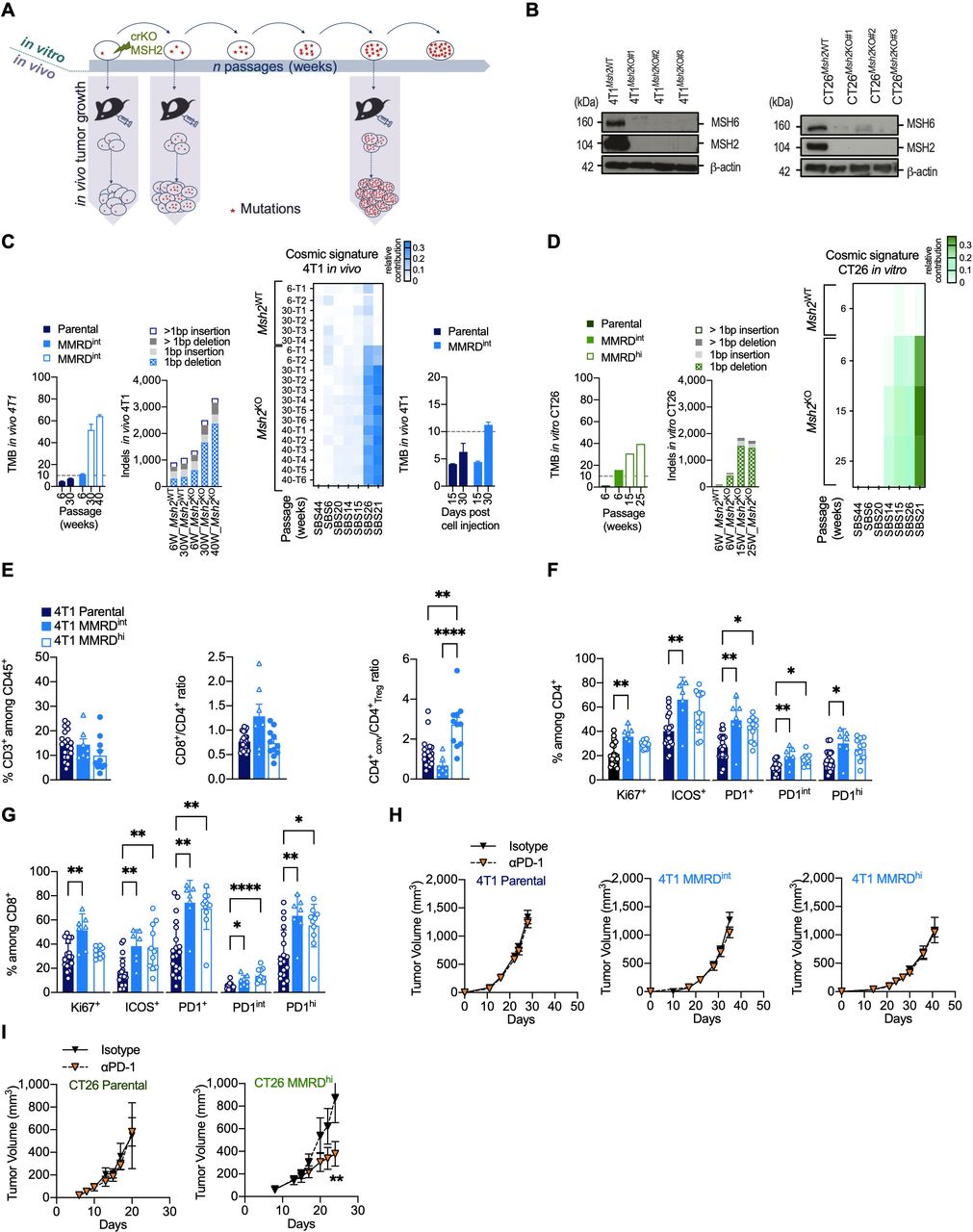 Figure 1
Figure 1 Variable responses to αPD-1 therapy in MMR-deficient mouse models. (A) Experimental model system for generating MMRD tumor cell lines (4T1, CT26) harboring increasing tumor mutation burden due to the inactivation of MSH2 gene. (B) Loss of MSH2 protein detected by western blot in clones inactivated for MSH2 gene by CRISPR/Cas9 in 4T1 (left panel) and CT26 (right panel). MSH6 which forms a heterodimer complex with MSH2 is destabilized in Msh2KO cells. (C–D) Counts of non-synonymous nucleotide substitution per Mb (TMB) and indels from whole-exome analysis in 4T1 and CT26 MSH2ko clones and in parental cells after serial passages (in weeks) in culture or tumor growth (D15, D30). Heatmap of relative contribution of MMR COSMIC signatures (signature 6, 14, 15, 20, 21, 26, 44) in different tumors injected at indicated passages (right panel). The TMB cut-off of 10 non-synonymous/Mb is represented in dotted lines. (E–G) Flow cytometry analyses of tumor-infiltrating leukocytes in 4T1 parental (dark blue), 4T1 MMRDint (p=6 weeks, blue) and 4T1 MMRDhi (p=30 weeks, white and blue) tumors resected 30 days post cell injection (n=7–20 tumors per group). The proportions of T lymphocytes (CD3+) among CD45+ cells, as well as the ratios CD8+/CD4+ and CD4+ conventional/Treg are depicted (E). Note that the Treg cell population was defined as CD4+ Foxp3+ cells and conventional CD4+ population was defined as CD4+ Foxp3– cells. The proportions of Ki67+, ICOS+ and PD1+ among CD4+ (F) and CD8+ cells (G) are shown. (H–I) Tumor growth kinetics of 4T1 (H) and CT26 (I) Parental (4T1 n=2, 12 mice/group; CT26 (n=1, 5–6 mice/group), MMRDint (4T1 n=3, 10–11 mice/group), and MMRDhi (4T1 n=2, 7–8 mice/group; CT26 (n=2, 12 mice/group) clones in mice treated with αPD-1 or its isotype when tumors reached 70–90 mm3. Data are shown as mean±SEM. Statistical analyses were performed using Mann-Whitney test when comparing two groups (H–I) and Dunn’s test (E–G) when comparing more than two groups. Symbol significance: *p≤0.05; **p≤0.01; ***≤0.001, ****≤0.0001. MMR, mismatch repair; MMRD, mismatch repair-deficient; MMRDhi, MMRD high (cells serially passaged for more than 15 weeks in culture); MMRDint, MMRD intermediate (cells serially passaged during 6 weeks in culture); PD-1, programmed death 1; TMB, tumor mutational burden (non-synonymous mutations/Mb); Treg, regulatory T cells.
Adoptive transfer of neutrophilsAdoptive transfer of neutrophils from naïve BALB/c mice or tumor-induced neutrophil (TIN) from 4T1 bearing BALB/c mice, was realized by retro-orbital injection of 106 cells. Untouched neutrophils or TIN were purified from spleens using the neutrophil isolation kit (Miltenyi order no. 130-097-658) according to manufacturer protocol. Cells were stained before and after enrichment with anti-Ly-6G and CD11b to control purity.
Immunohistochemical analysis of mice tumor tissueAll immunohistochemical techniques were performed on an automated stainer (Bond RX, Leica Biosystems, Nanterre, France). For detection of CD8, deparaffinized 3 µm-thick tissue sections were submitted to heat-induced antigen retrieval for 20 min at 100°. Sections were then incubated with the primary antibodies (CD8 clone D4W2Z, Cell Signaling, Danvers, Massachusetts, USA) for 1 hour at room temperature. The signal was revealed with 3, 3'-diaminobenzidine (DAB). For simultaneous detection of CD4 and FoxP3, after antigen retrieval, sections were successively incubated with the primary antibodies (CD4 rabbit monoclonal D7D2Z, Cell Signaling and anti-FoxP3 rabbit monoclonal antibody D608R, Cell Signaling), each for 1 hour at room temperature, detected respectively with Bond Polymer Refine Red Detection (Leica Biosystems) and Bond Polymer Refine Detection kit (Leica Biosystems). Sections were successively revealed by Red chromogen (Leica Biosystems) and HIGHDEF Black HRP chromogen/substrate (Enzo Life Sciences, Villeurbanne, France).
PatientsAll patients included in this study had previously received at least one line of therapy. They were enrolled from November 2014 to October 2020 at Gustave Roussy and provided informed consent before enrollment. The study is reported in Health Data Hub. Information regarding Eastern Cooperative Oncology Group status, National Clinical Trial (NCT) number, inclusion/exclusion criteria, line of treatment are indicated in online supplemental table S2. White blood cell (WBC) count, absolute lymphocyte count (ALC), absolute neutrophil count (ANC), serum lactate dehydrogenase, and other clinical data, including metastasis (number and site), immunohistochemistry results (for MSI diagnostic confirmation), and response) were collected before starting αPD-L1 treatment and until last follow-up. Patients were treated with αPD-L1 until disease progression, intolerable toxicity and/or the investigator’s decision. Tumor assessment was performed at baseline, and during treatment with varying frequency. Clinical response was classified according to Response Evaluation Criteria in Solid Tumors (RECIST). The Royal Marsden Hospital Prognostic Score was calculated using the formula described by Garrido-Laguna et al.23
NLR was calculated as follows: NLR=ANC/ALC from peripheral blood of patients. NLR change (NLRchange) was calculated as follows: ((NLRearly on-treatment – NLRbaseline)/NLRbaseline)×100. Note that NLR change less than 0% means lymphocytes are becoming more abundant relative to neutrophils. Progression-free survival (PFS) was determined from the first cycle of treatment to disease progression documented by imaging, or death (event), or last follow-up (censored). Overall survival (OS) was calculated from the first cycle of treatment to death (event) or last follow-up (censored). The objective response rate (ORR) was defined as the number of patients with either a complete response (CR) or a partial response (PR) divided by the number of patients who were treated in the study.
Immunohistochemical analysis of human tumor tissuesImmunohistochemical techniques were applied to 3 µm-thick deparaffinized sections of Formalin-Fixed, Paraffin-Embedded (FFPE) tissues and performed on automated stainers, after heat-induced antigen retrieval. CD15 was detected with the mouse monoclonal antibody MMA (Roche Diagnostics, Basel, Switzerland) and CD3 with a rabbit polyclonal antibody (DAKO, Glostrup, DK). CD3, CD15, CD15/CD3 change were, respectively, calculated as follow: (CD3/mm²during SD or progression – CD3/mm²before ICB initiation)/CD3/mm²before ICB initiation))×100; : (CD15/mm²during SD or progression – CD15/mm²before ICB initiation)/CD15/mm²before ICB initiation))×100; : (CD15/CD3during SD or progression – CD15/CD3before ICB initiation)/ CD15/CD3before ICB initiation))×100.
Statistical analysisData analyses were performed using Prism V.9 (GraphPad, San Diego, California, USA). Data are shown as mean±SEM. At the same time point, statistical analyses were performed using Mann-Whitney test when examining two groups and Dunn’s or regular one-way analysis of variance test when examining more than two groups. At different time points, statistical analyses were performed using Fishers Least Significant Difference (LSD) Fisher’s test. Statistical analysis for fraction survival using Kaplan-Meier estimation curves, were performed by using log-rank (Mantel-Cox) test and HR values were assessed with Mantel-Haenszel test. Symbol significance: *p≤0.05; **p≤0.01; ***p≤0.001, ****p≤0.0001.
ResultsGenetic and immune characterization of MMRD murine tumor modelsTo generate MMRD murine models, we used CRISPR/Cas9-mediated genetic editing to inactivate the Msh2 gene in breast (4T1) and colorectal (CT26) murine cancer cell lines (figure 1A,B). The 4T1 tumor cell line was chosen for its very low background mutation rate.24 Moreover, when injected into the mammary fat pad of BALB/c mice, 4T1 cells are refractory to αPD-1 or αCTLA-4 and are associated with an accumulation of neutrophils and other granulocytic cells.24 25 In contrast, CT26 tumor cells contain a substantial number of somatic mutations. When transplanted subcutaneously into the right flank in BALB/c mice, CT26 have been shown to respond to αCTLA-4 but not to αPD-1, and is not associated with neutrophil accumulation.24 26
Given that mutations accumulate in each round of DNA replication in MMR-deficient cells, parental cells and MMRD derivatives were serially passaged in vitro, and TMB as well as indels were monitored over time in both 4T1 and CT26 MMRD models (figure 1C,D). We observed a gradual increase in TMB in 4T1Msh2KO tumors with passage, reaching up to 64 mutations/Mb at passage 40 weeks. As expected, TMB remained stable in 4T1Msh2WT tumors over time (figure 1C). The mutagenic spectrum caused by Msh2 inactivation was consistent with COSMIC mutational signatures associated with MMR deficiency (ie, signatures 6, 14, 15, 20, 21, 26, and 44) with a relatively high contribution of signatures 21 and 26 in 4T1Msh2KO 2. We also found an important increase in indels in 4T1Msh2KO cells after 40 weeks in cell culture, with prominent single base pair deletions (−1 bp). To monitor the accumulation of mutations during tumor growth, 4T1Msh2KO cells at passage 6 weeks were transplanted into mammary fat pads in BALB/c mice, and tumor exomes at two time points post-injection were analyzed (figure 1C, right panel). We observed an increase in TMB in 4T1Msh2KO cells between Day 15 and Day 30 (4.5 vs 11.25 mut/Mb), while in 4T1 parental cells, TMB remained relatively stable over time (4.2 vs 6.3 mut/Mb). These genetic features were corroborated in the MMRD CT26 syngeneic model (figure 1D). Altogether, these results indicate that our two MMRD murine models recapitulate the mutational landscape found in human tumors with MSI. MMRD cells passaged for 6 weeks are hereafter referred to as MMRDint cells, and MMRD cells that were passaged for more than 15 weeks are referred to as MMRDhi cells.
A high density of activated tumor-infiltrating lymphocytes (TILs) has been widely described in MSI tumors in the clinic.4 27 28 MMR deficiency in CT26 model has been shown to cause a slight increase in immune infiltration compared with parental cells29 while immune features in 4T1 MMRD models are unknown. Therefore, we investigated the TME in 4T1 MMRD tumors. TME was evaluated 30 days post injection for the parental, MMRDint and MMRDhi 4T1 (figure 1E,G). No difference was observed for CD3+ T cell infiltration. Interestingly in MMRDhi tumors, the conventional CD4+ T to Treg cell ratio (CD4+conv/CD4+Treg) was significantly increased compared with that in parental and MMRDint tumors (figure 1E, right panel), suggesting a less immunosuppressive microenvironment only in highly mutated tumors. MMRDint tumors exhibited higher proliferation (ie, Ki67+) of TILs compared with parental tumors, which was more pronounced within the CD8+ T cell population (figure 1F,G), consistent with the higher CD8+/CD4+ ratio in this model (figure 1E, right and middle panels). The inducible T cell co-stimulator (ICOS) activation marker and PD-1 expression were significantly increased within the CD4+ T and CD8+ T cell populations in both MMRDint and MMRDhi tumors compared with parental tumors (figure 1F,G). Altogether, these data indicate a higher activation of TILs in 4T1 MMRD tumors compared with parental tumors.
αPD-1 treatment is not effective in all MMRD modelsConsidering the immune infiltration in both MMRD CT26 and 4T1 tumors29 and this study, respectively, we anticipated that these murine models would respond to αPD-1 therapy. As expected, the parental tumors did not respond to αPD-1 therapy (figure 1H, left panel and figure 1I, left panel). αPD-1 monotherapy had no effect in MMRDint and MMRDhi 4T1 tumors (figure 1H, middle and right panels), while a significant reduction in tumor growth was observed in the MMRDhi CT26 tumor model (figure 1I, right panel). This result was not anticipated since these three MMRD models have a high TMB (CT26 MMRDhi TMB>30 mut/Mb; 4T1 MMRDint TMB>10 mut/Mb; and 4T1 MMRDhi TMB>50 mut/Mb) and a substantial immune infiltration with a strong PD-1 expression. Thus, despite the presence of clinically validated biomarkers for predicting response to αPD-1, treatment was ineffective in MMRD 4T1 models. This outcome suggests that other mechanisms are involved in primary resistance, even in highly mutated tumors.
Neutrophils are associated with αPD-1 resistance in MMRD tumorsContrary to the CT26 tumor model, 4T1-bearing mice have a massive increase in the number of circulating WBCs relative to normal control hosts; myeloid cells such as neutrophils largely account for this expansion of the WBC compartment, which might contribute to ICB resistance in parental 4T1 cells.30 However, this phenomenon remains unstudied in MMRD tumor models. We thus monitored the myeloid cell to lymphocyte (myelo/lympho) ratio in the blood of mice after transplantation of parental and MMRD tumor cells. We observed a 10-fold to 20-fold increase in the myelo/lympho ratio in the blood of parental and MMRDhi 4T1 tumor-bearing mice as compared with CT26 model (figure 2A). Similarly, a 5-fold to 15-fold increase in the myelo/lympho ratio was detected in the TME of both parental and 4T1 MMRDhi tumor models. Hence, the increase in indels and single base substitutions in MMRD 4T1 cells did not prevent the accumulation of myeloid cells in these preclinical tumor models.
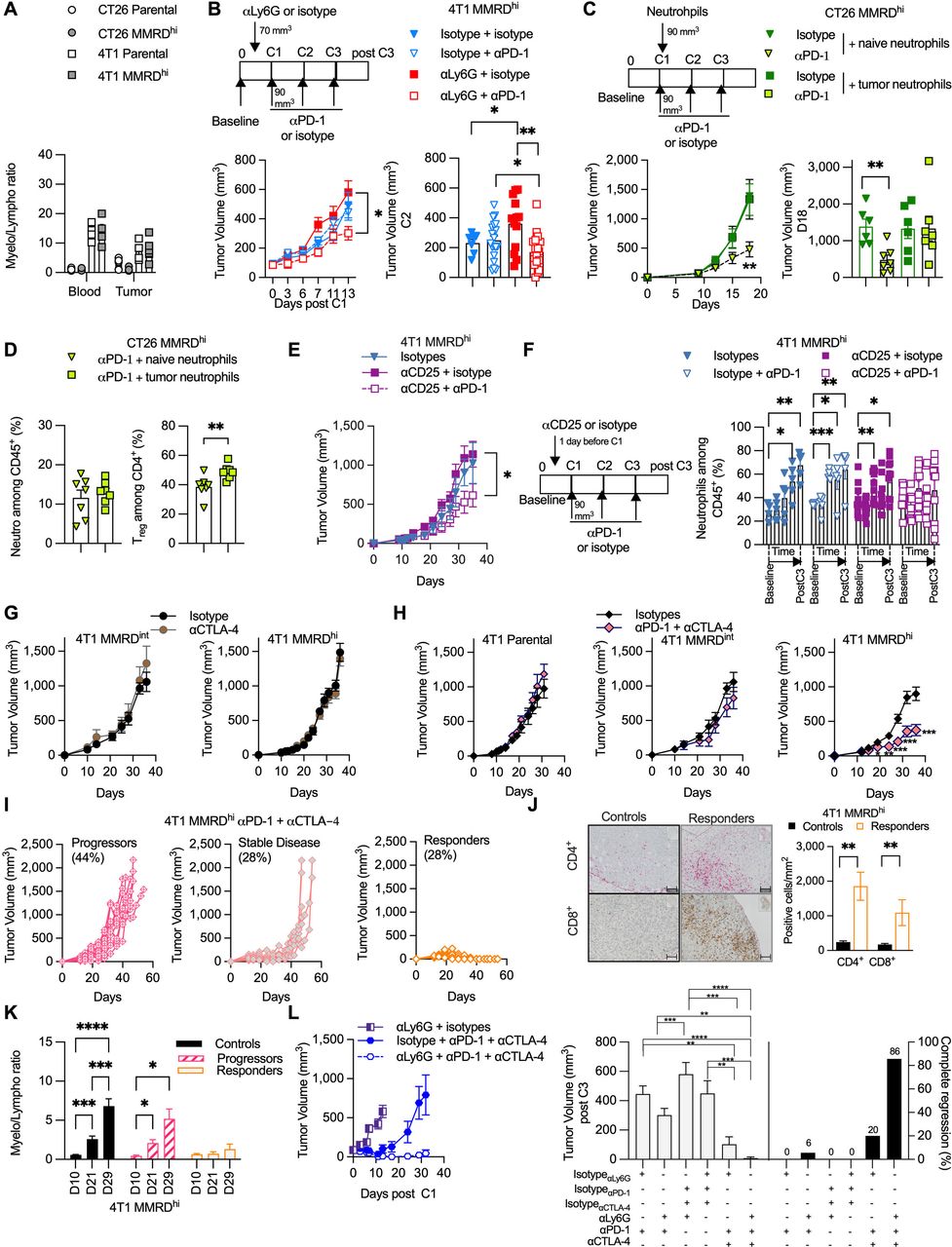 Figure 2
Figure 2 Neutrophil levels negatively correlate with αPD-1 response in MMRD preclinical tumor models. (A) Analysis of blood myeloid ratio (defined as the ratio of myeloid cells/lymphocytes, both gated among CD45+) in mice injected with 4T1 parental (white square), 4T1 MMRDhi (gray square), CT26 parental (white circle), CT26 MMRDhi (gray circle) (n=1, 4–6 mice/group). (B) Tumor growth kinetics of 4T1 MMRDhi mice±depleted for neutrophil cells using αLy6G antibody (clone 1A8) and subsequently treated with αPD-1 or its respective isotype during 13 days post first injection of αPD-1 (n=3, 7–20 mice in each group). Scheme of experimental design is shown (upper panel). Tumor volume just before the second αPD-1 injection is shown (lower right panel). (C) Mice bearing CT26 MMRDhi cells were injected with 250,000 neutrophils originated from either naïve mice or mice bearing 4T1 tumors. Mice were concomitantly treated with αPD-1 or its isotype and tumor growth was monitored for each group (n=1, 6–7 mice/group). Scheme of experimental design is shown (upper panel). Tumor volume at Day 18 post cell injection is shown (lower right panel). (D) Flow cytometry analyses of tumor neutrophils (gated as CD11b+ CD11c– SiglecF– Ly6Ghi Ly6Cint, among CD45+) and Treg cells (gated as CD4+ Foxp3+ among CD4+) in CT26 MMRDhi treated with αPD-1 and injected with 250,000 neutrophils originated from either naïve mice (green triangle) or mice bearing 4T1 tumors (green square) (n=7 mice in each group). (E–F) Tumor growth kinetics (E) and longitudinal flow cytometry analyses of blood neutrophils (F) of 4T1 MMRDhi mice±depleted for Treg cells using αCD25 (PC-61.5) antibody and subsequently treated with αPD-1 or its respective isotype over a 35-day period (n=2, 5–10 mice in each group). (G) Tumor growth kinetics of 4T1 MMRDint (n=1, 5–6 mice/group) and MMRDhi clones (n=1, 7–8 mice/group) in mice treated with αCTLA-4 or their isotypes when tumors reached 70–90 mm3 (6–25 mice/group). (H) Tumor growth kinetics of 4T1 parental (n=1, 6 mice/group), MMRDint (n=1, 6 mice/group), and MMRDhi clones in mice treated with αCTLA-4+αPD-1 or their isotypes when tumors reached 70–90 mm3 (n=4, 22–25 mice/group). (I) Spider plots of individual tumor growth of MMRDhi tumor-bearing mice (passage 40 weeks) treated with αPD-1+αCTLA-4 over a 45-day period. (J) Representative photographs of CD4+ and CD8+ immunohistochemistry staining of 4T1 MMRDhi tumor-bearing mice 2–3 days after the third dose of αPD-1+αCTLA-4 or their respective isotypes. Histogram of the density of CD4+ and CD8+ cells in indicated groups (n=1, 6 mice/group). Scale bars=50 µm. (K) Blood myeloid ratio was evaluated at D10 (before first dose of ICB), D21, D29 post cell injection in mice treated with isotypes (black) or in mice that progressed (pink stripes) or responded (orange) to αPD-1+αCTLA-4 treatment (n=1, 6–10 mice/group) (L) Tumor growth kinetics of 4T1 MMRDhi in mice±depleted for neutrophil cells using ɑLy6G (1A8) antibody and treated with αPD-1+/–αCTLA-4 or their respective isotypes (n=1, 5–12 mice in each group). Tumor volume 3 days after the last administration of immune checkpoint blockade is shown for each group with the percentage of complete tumor regression. Data are shown as mean±SEM. At the same time point, statistical analyses were performed using Mann-Whitney test when examining two groups (D, J) and Kruskal-Wallis or regular one-way analysis of variance test (L) when examining more than two groups. At different time points, statistical analyses were performed using Fisher LSD test (B, E–H, K). Symbol significance: *p≤0.05; **p≤0.01; ***≤0.001, ****≤0.0001. CTLA-4, cytotoxic T-lymphocytes-associated protein 4; MMRD, mismatch repair-deficient; MMRDhi, MMRD high cell; PD-1, programmed death 1; Treg, regulatory T cells; Fishers Least Significant Difference (LSD)
To determine whether neutrophils preclude an αPD-1 response in MMRDhi 4T1 tumors, mice were treated with the neutrophil-depleting antibody αLy-6G31 32 or its isotype control. As expected, αLy6G -treated tumor-bearing mice showed a reduction in circulating neutrophil numbers compared with mice treated with isotype control (online supplemental figures S1 and S2). While αLy6G did not significantly modify the tumor growth of MMRDhi 4T1 tumors, αPD-1 treatment along with αLy6G induced significant reduction of the tumor growth in MMRDhi 4T1 tumors (figure 2B). To demonstrate the direct role of TIN, we adoptively transferred neutrophils from non-tumor-bearing BALB/c mice or neutrophils from 4T1 tumor-bearing mice into the mice bearing MMRDhi CT26 tumors, sensitive to αPD-1 treatment. As shown in figure 2C, adaptive transfer of TIN abrogated the efficacy of αPD-1 in MMRDhi CT26 tumors. No such effect was observed with naïve neutrophils (figure 2C) demonstrating that only TIN hampered αPD-1 efficacy. We next examined the TME of MMRDhi CT26 tumors after adoptive transfer of naïve neutrophils or TIN. Surprisingly, while no difference in neutrophil proportion was observed, Treg cells were significantly increased in the TME when TIN were adoptively transferred (figure 2D), suggesting that TIN favored Treg infiltration and/or amplification.
Neutrophils and Treg cells are relatedWe next decided to deplete the Treg cell population in 4T1 tumor-bearing mice with a CD25-depleting antibody. Depletion with an αCD25 antibody is not straightforward, as Treg depletion before tumor implantation induces total tumor regression in approximately 50% of mice.33 Thus, the injection of the αCD25 antibody was performed 1 day before αPD-1 treatment, resulting in a decrease in Treg cells in the blood (online supplemental figure S3 upper panel).
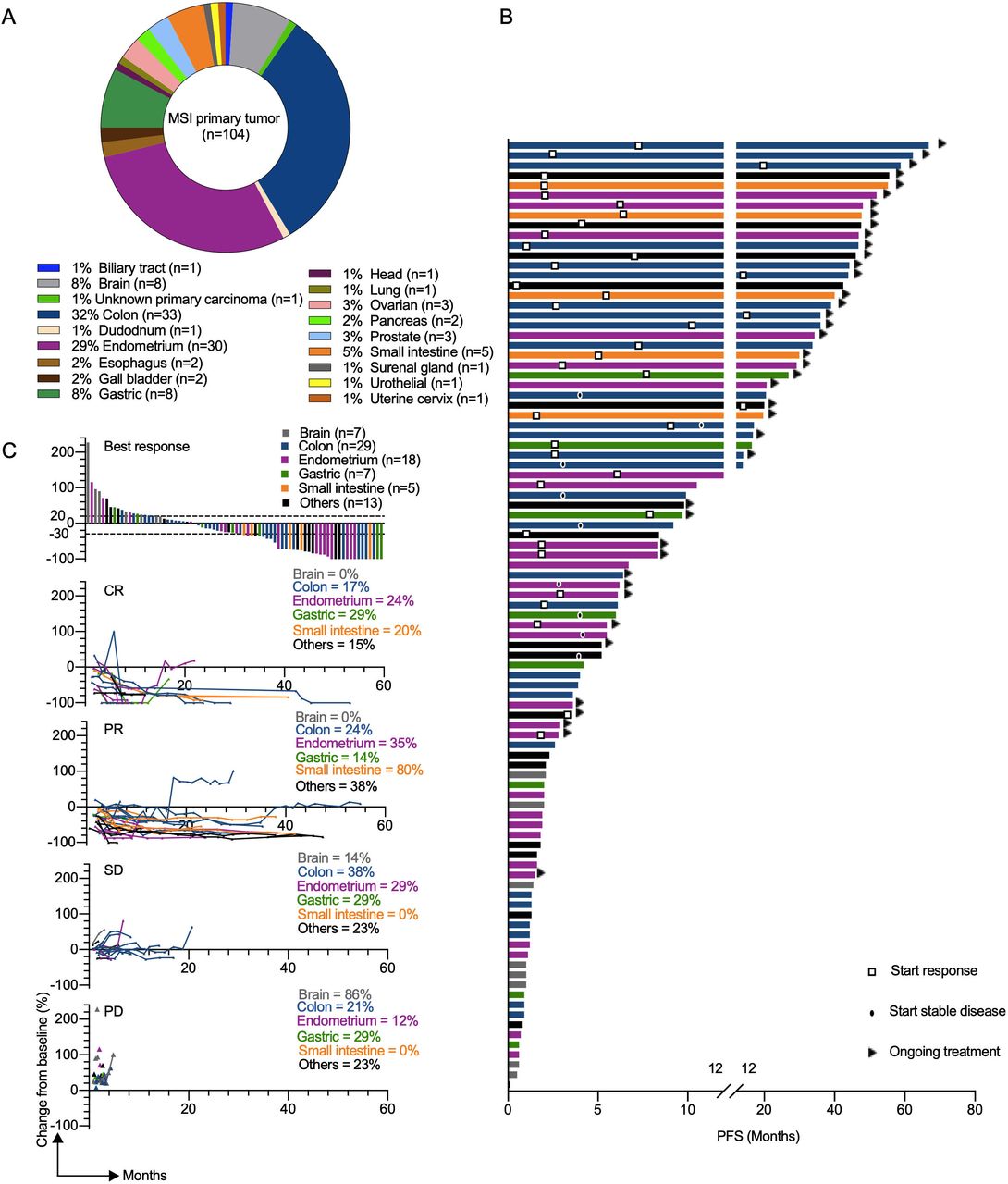 Figure 3
Figure 3 Clinical response to αPD-L1 in patients with different MMRD tumor types. (A) Pie chart representing classification of MSI/ MMR-deficient tumor types analyzed in the GR cohort. (B) the swimmer plots illustrate the PFS of patients treated with αPD-L1 antibody. Ongoing treatment is represented with a triangle and the start of response or stable disease was tagged by a square or a black oval, respectively. Histological tumor types are color coded. (C) Waterfall plot of best response (defined as best percentage change from baseline in tumor size by RECIST V.1.1) to αPD-L1 therapy (upper panel). Spider plots (defined as longitudinal tumor growth or shrinkage compared with baseline accorded to RECIST V.1.1) in the cohort (lower panels). Analysis included all patients who received at least one dose of study treatment and had at least one evaluable post-baseline tumor assessment. CR, complete response; MSI, microsatellite instability; MMR,mismatch repair; MMRD, mismatch repair-deficient; PD, progressive disease; PD-L1, programmed death ligand 1; PFS, progression-free survival; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease.
Moreover, even if αCD25 therapy is associated with a potential risk of depletion of CD25+ T effector cells and not only Treg cells, we did not observe a clear difference in the CD4+ cell proportion between all groups (online supplemental figure S3 lower panel). As anticipated, αPD-1 treatment along with Treg cell depletion restored antitumor activity in the MMRDhi 4T1 model (figure 2E). Interestingly, no accumulation of neutrophils was observed in mice treated with αCD25+αPD-1 in contrast to mice in the other groups (figure 2F). These data suggest that immunoregulatory effectors such as neutrophils and Treg cells appear to be related and counter αPD-1 efficacy even in highly mutated MMRDhi 4T1 tumors.
Given the widely described role of αCTLA-4 in modulating Treg cell suppressive functions,34 αCTLA-4 +/– αPD-1 was administered. Mice transplanted with MMRDint or MMRDhi 4T1 cells were resistant to αCTLA-4 monotherapy (figure 2G). However, combination treatment with αCTLA-4+αPD1 triggered tumor shrinkage in MMRDhi 4T1-bearing mice, while parental and MMRDint tumors were still unresponsive (figure 2H). Accordingly, survival was increased in MMRDhi 4T1-bearing mice treated with combination treatment (online supplemental figure S4). By performing rechallenge experiments, we demonstrated that mice curing MMRDhi primary tumors with the αCTLA-4 + αPD-1 combination were protected from MMRDhi 4T1 tumors but not from parental 4T1 tumor cells, although we observed a tumor growth delay (online supplemental figure S5). These results suggest that the combination of αPD-1 and αCTLA-4 blockade elicits an effective and long-lasting memory immune response in MMRDhi 4T1 tumors, possibly directed against neoantigens.
 Figure 4
Figure 4 NLRchange can predict resistance to αPD-L1 therapy for patient with different MMRD tumor types.
留言 (0)