
記住我





Historically, mesenchymal tumors have been defined on the basis of morphological features that correspond to an apparent line of differentiation, such as adipocytic or smooth muscle differentiation. Advances in immunohistochemistry helped to confirm this line of differentiation in these well established entities, but also broadened the morphological spectrum of these tumors, as the resemblance to the normal histological counterpart is not always overt. However, in some tumors aforementioned features are not sufficient to define these tumors in a proper manner, as these have undifferentiated or poorly differentiated morphology or the line of differentiation is uncertain. Especially in this group of tumors, the advances of molecular techniques allowing the elucidation of numerous genetic alterations have been irrefutably important in their classification. This is clearly reflected in the current fifth edition of the WHO Classification of Soft Tissue and Bone Tumors [1], where the integration of molecular findings has become increasingly important, sometimes so far-reaching that some entities are currently defined by and named after their genetic alteration. In the previous fourth edition of the WHO, undifferentiated small round cell sarcoma other than Ewing sarcoma consisted of an uncharacterized group of small blue round cell sarcomas lacking the canonical Ewing fusion involving one member of the FET family and a member of the ETS family. Over the past decade, increasing evidence supported the notion that this group of ‘Ewing-like’ sarcomas included a few specific subgroups characterized by specific molecular alterations [2▪▪,3▪▪]. This has led to the recognition of three new distinct entities in the category of undifferentiated small round cell sarcoma of bone and soft tissue, named after their molecular alteration: ‘round cell sarcoma with EWSR1-non-ETS fusions’, ‘CIC-rearranged sarcoma’, and ‘sarcoma with BCOR genetic alteration’ [1]. The delineation of these entities based on their underlying molecular profile has enabled the identification of specific molecular markers for these entities and has even revealed subtle differences in morphology [2▪▪,3▪▪]. There are different viewpoints as to whether the demonstration of the specific molecular alteration for an entity that includes the alteration in its name is required, or essential, or only ‘desirable’ as is the current consensus in the WHO classification. Nevertheless, the distinction between the different types of round cell sarcomas is important since the biological behavior and the response to chemotherapy differ between these entities [3▪▪].
 Box 1:
Box 1: no caption available
Similarly, molecular genetics have a pivotal role in a group of tumors that is provisionally included in the fifth edition of the WHO classification, including NTRK-rearranged spindle cell neoplasm and EWSR1-SMAD3-positive fibroblastic tumors. In general, these tumors are rare, have a broad morphological spectrum and clinical behavior and an unspecific immunophenotype, but are unified by the presence of a shared fusion [1,4,5]. In these entities, the molecular finding is leading in the diagnosis and therefore molecular studies are required for a definite diagnosis. Currently, these emerging entities still need to be better defined.
In other newly introduced entities, molecular alterations have played a complementary role to conventional histology and immunohistochemistry in their delineation. Most of these tumors were already recognized to be distinct from existing entities by specific clinical and pathological features, but the identification of specific molecular findings has contributed to separate them from other entities. This review will discuss these newly introduced and emerging bone and soft tissue entities that are either defined by their molecular hallmark or in which the molecular findings have played a role in their recognition as a separate entity.
NEW ENTITIES DEFINED BY A MOLECULAR ALTERATIONRound cell sarcomas with EWSR1-non-ETS fusion are malignant tumors composed of undifferentiated round and/or spindle cells in which the EWSR1/FUS fusion partner does not belong to the ETS family of transcription factors. PATZ1 and NFATC2 are most commonly involved and their clinical features and morphology are different. EWSR1-NFATC2 sarcomas occur predominantly in bone [6], while FUS-NFATC2 sarcomas are exclusively reported in the long bone. Both occur mostly in children and adults with a male predominance [7]. Morphologically, the cells in EWSR1/FUS-NFATC2 rearranged sarcomas may form nests, trabeculae and cords with a variable amount of extracellular matrix. Half of the cases show diffuse expression of CD99 and variable expression of keratin AE1/AE3 and CD138 have been reported [7]. Data on clinical outcome are still sparse and heterogenous, as some tumors behave highly aggressive and some patients are cured after complete resection, but with the potential for late metastasis [2▪▪,3▪▪]. Of note, identical EWSR1-NFATC2 rearrangements have been recently described in a spectrum of benign vascular lesions of bone, including a hemangioma/vascular malformation-like lesion and an unusual benign epithelioid vascular tumor, as well as in simple bone cyst. The amplification of the fusion product that is characteristic of EWSR1-NFATC2-rearranged sarcoma is absent in these benign NFATC2 rearranged lesions and there is no morphological overlap [8–10]. These findings illustrate that molecular findings should always be interpreted within the proper morphological, immunohistochemical, and clinicoradiological context. EWSR1-PATZ1 sarcomas have a wide age distribution, without gender predominance and typically arise in the deep soft tissue. Data from the largest clinicopathological study so far, which included nine EWSR1-PATZ1 sarcomas, suggest that a subset might have a more indolent behavior than previously appreciated [11].
CIC-rearranged sarcoma is a high-grade undifferentiated round cell sarcoma, in which CIC is most often (95%) fused with DUX4. Other gene partners include FOXO4, LEUTX, NUTM1, and NUTM2A. They occur most commonly in young adults within the somatic soft tissues and have a wide spectrum of morphology including round, epithelioid and spindle cells with frequent myxoid stromal changes. In addition to CD99 and WT-1, ETV4 is commonly expressed and can be used as a diagnostic marker, as the fusion leads to enhanced CIC-transcriptional activity and therefore upregulation of ETV4 among other genes. In CIC-NUTM1 fused sarcomas, NUT is also immunohistochemically overexpressed. CIC-rearranged sarcomas behave aggressively with an inferior overall survival compared with Ewing sarcoma [12].
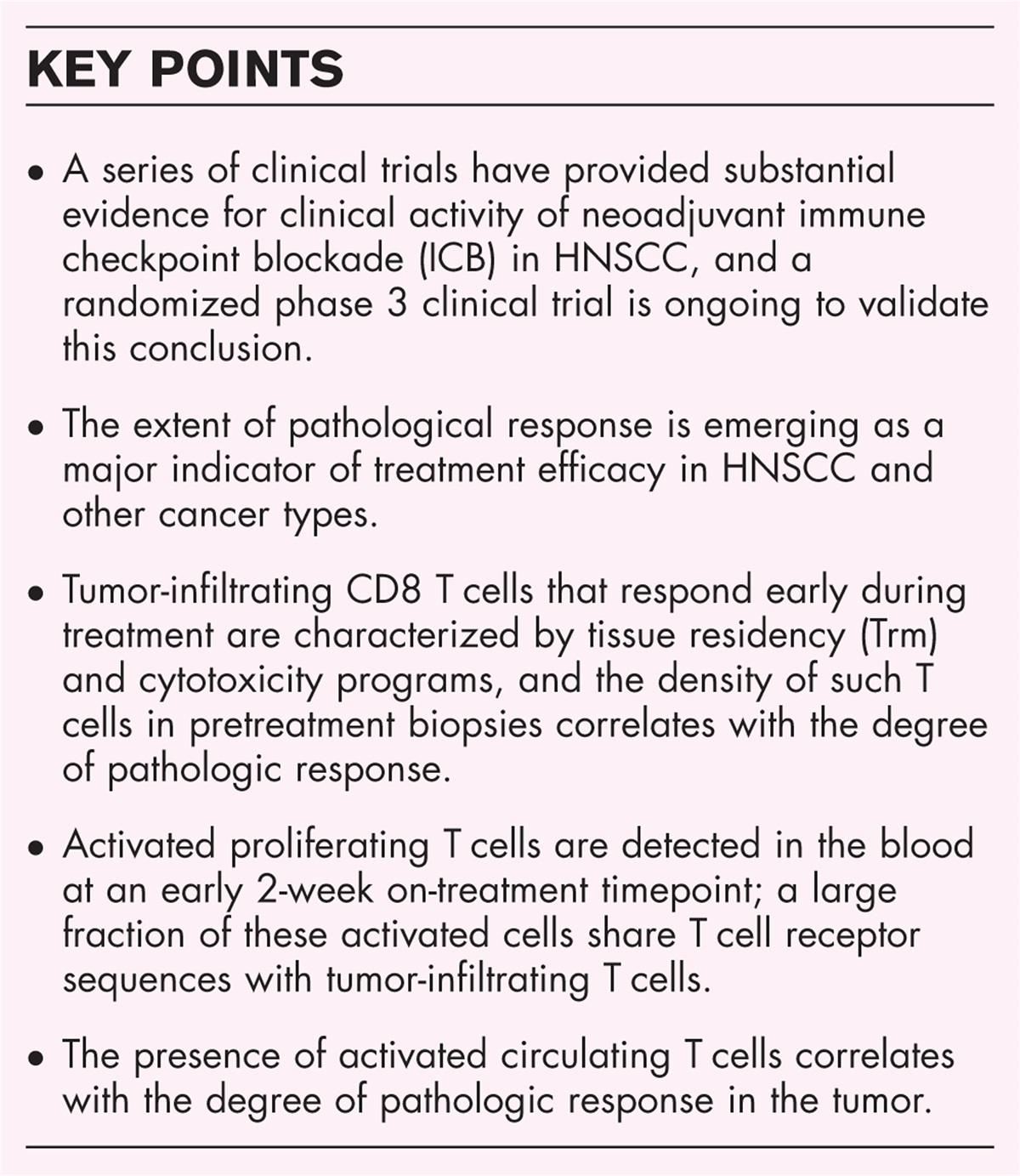
Sarcoma with BCOR genetic alteration includes two tumor groups unified by the overlapping morphology, immunohistochemical features and genotype, resulting in overexpression of BCOR, though with distinct clinical characteristics. One group is characterized by sarcomas with a BCOR-fusion, most often involving BCOR-CCNB3 and less frequently BCOR-MAML3 and ZC3H7B-BCOR[13,14]. BCOR-CCNB3-positive tumors have a strong male predominance and occur most often in the bones of patients younger than 20 years old. This stands in contrast to alternative BCOR-rearranged sarcoma, which is seen in a wider age range and affects both sexes equally [13–15]. The other group is characterized by BCOR internal tandem duplications, occurring in infantile undifferentiated round cell sarcoma and primitive myxoid mesenchymal tumor of infancy [16]. Tumor cells show diffuse nuclear expression of BCOR, SATB2, TLE1, cyclin D1, and CD99 in the majority of the cases (Fig. 1). The outcome of the BCOR family tumors is not well studied, although it is known that it shows survival rates similar to Ewing sarcoma and a favorable response to Ewing sarcoma chemotherapy regimens. However, a substantial proportion of patients develop lung metastasis [15,17].
 FIGURE 1:
FIGURE 1: Sarcoma with BCOR genetic alteration. H&E shows small cells with round to oval nuclei (a). In contrast to Ewing sarcoma CD99 shows patchy staining instead of a typical honeycomb membranous staining (b). BCOR is strongly expressed in the nuclei (c), as it is upregulated due to the BCOR-CCNB3 fusion, confirmed by targeted next-generation sequencing (d).
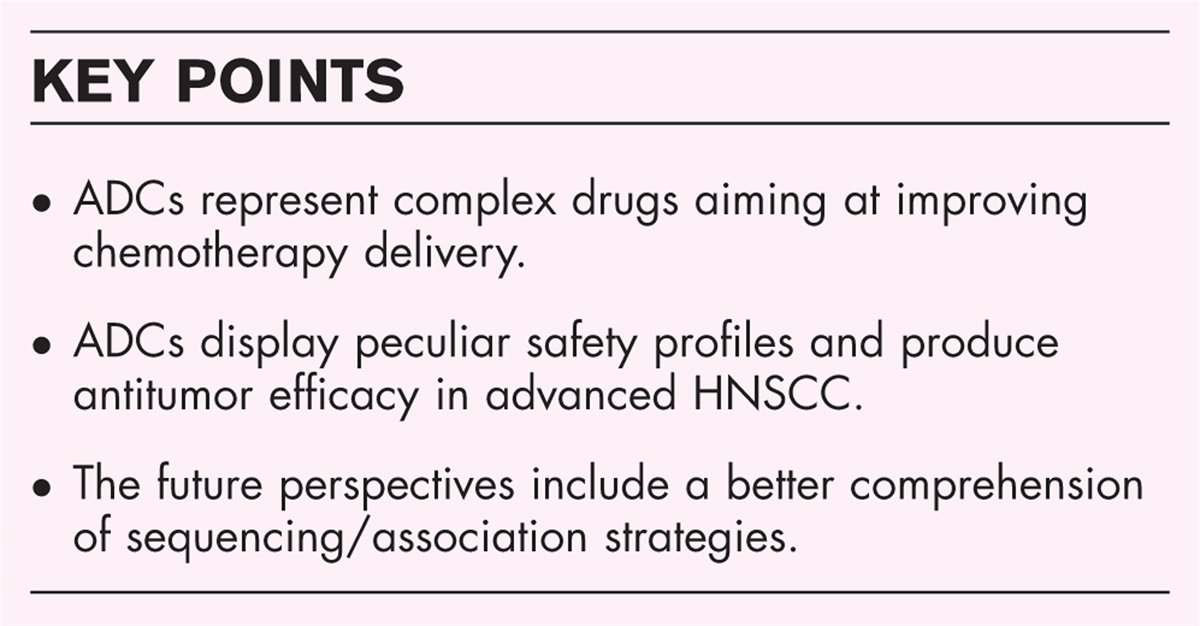
EMERGING ENTITIES DEFINED BY A MOLECULAR ALTERATIONEWSR1-SMAD3-positive fibroblastic tumor is the provisional nomenclature for this entity first described in 2018 by Kao et al.[18]; RNA-sequencing revealed a novel EWSR1-SMAD3 gene fusion in a nonclassifiable spindle cell neoplasm arising in the heel of an infant. The majority involve superficial tissue and are located in acral locations in women. Immunohistochemically, the neoplastic cells consistently express ERG, without expression of SMA, CD34, CD31, and S100 (Fig. 2) [18–20]. Although it is considered benign, with local recurrence if not completely excised, further case series are awaited to better delineate the specific clinical presentation and behavior of this emerging entity.
 FIGURE 2:
FIGURE 2: EWSR1-SMAD3-positive fibroblastic tumor. H&E shows a bland spindle cell proliferation (a) with nuclear expression of ERG, as is consistently seen in these tumors (b). Targeted next-generation sequencing fusion analysis reveals a fusion between EWSR1 and SMAD3 (c).
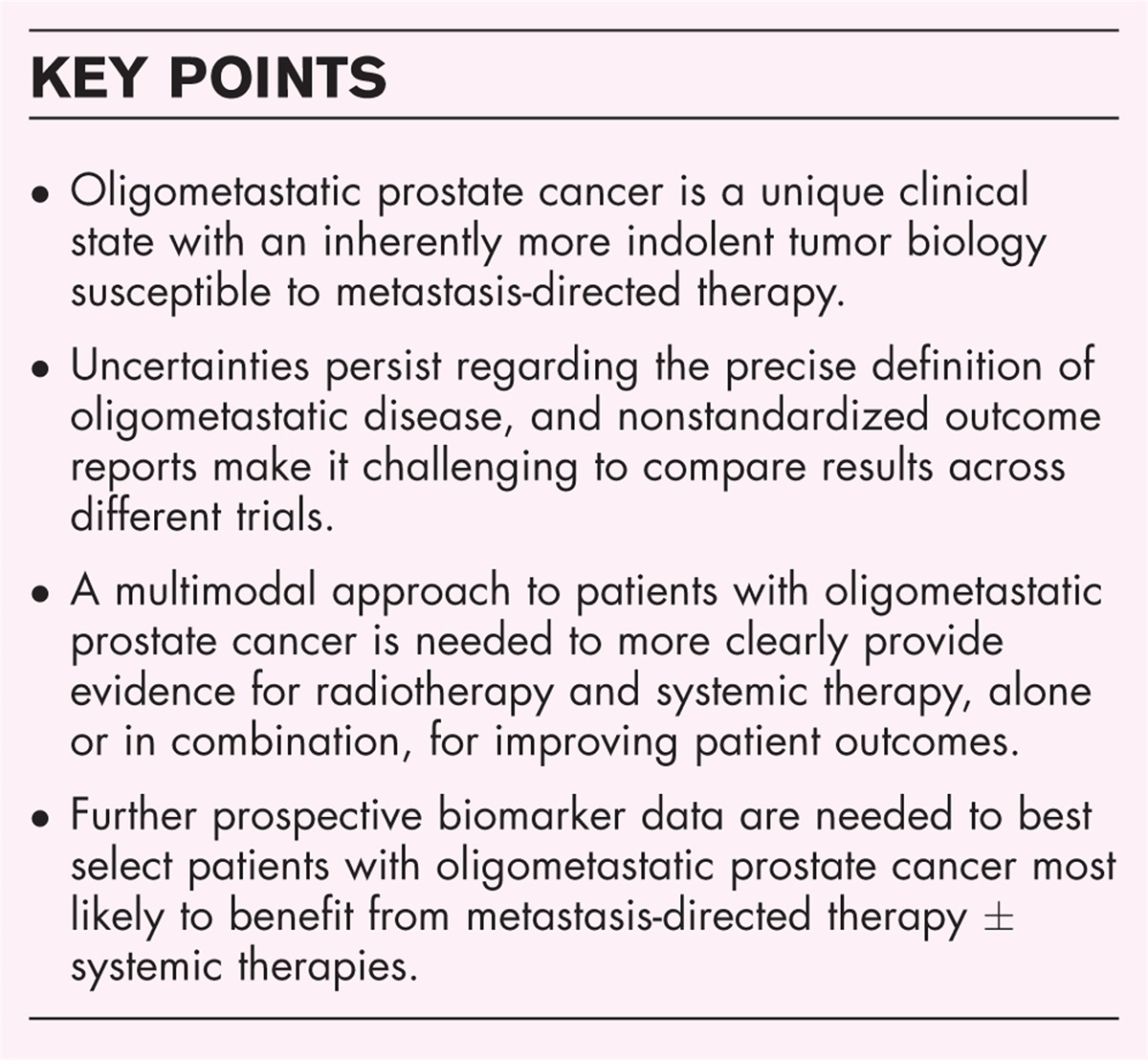
NTRK-rearranged spindle cell neoplasm is another emerging entity that includes a group of spindle cell neoplasms defined by NTRK fusions (Fig. 3). These fusions are not solely confined to this specific group, as NTRK fusions have been reported at high frequency in different cancers, such as infantile fibrosarcoma and secretory breast carcinoma, as well as in a low frequency in various other well established tumors, such as gastrointestinal stromal tumor [5,21]. The group of NTRK-rearranged spindle cell neoplasms represents a very heterogeneous group of tumors with a wide morphological spectrum and variable clinical presentation, unified by a shared gene fusion [5,22]. The prognosis is highly related to the morphological phenotype, varying between an indolent course to distant metastasis and death from disease [4,22]. Most tumors harbor a fusion involving NTRK1 and to a lesser extend NTRK2 and NTRK3 and are reactive for antipan-TRK immunohistochemistry (Fig. 3). As NTRK-fused tumors are targetable with TRK-inhibitors, this offers patients a potential treatment option. Alternative kinase fusions involving RAF1 and BRAF are described in tumors with identical morphologic and immunophenotypic features [4].
 FIGURE 3:
FIGURE 3: NTRK-rearranged spindle cell neoplasm. T1-weighted MRI after Gadolinium contrast showing a destructive tumor in the pelvis with a large soft tissue component (a), with a major response after treatment with a tyrosine kinase inhibitor and radiotherapy (b). Gross evaluation showing myxoid changes in the soft tissue, no evident tumor mass is visible (c). H&E of biopsy previous to treatment showing atypical stellate spindle cells in a myxoid background (d) with immunohistochemical expression of pan-TRK (insert). Next-generation sequencing fusion analysis reveals a fusion between CNTN4 and NTRK3 (e).
NEW ENTITIES SUPPORTED BY A MOLECULAR ALTERATIONAtypical spindle cell / pleomorphic lipomatous tumor was initially described as a morphological variant of well differentiated liposarcoma (WDLPS) by Dei Tos et al. and was designated as ‘spindle cell liposarcoma’. However, it has been revealed that these tumors behave as benign lesions with the tendency for local recurrence which may require surgical excision with clear margins [23]. These tumors are poorly marginated, have a predilection for the limbs and limb girdles and arise in the subcutis. Morphologically, the tumor is composed of atypical spindle cells, adipocytes and (pleomorphic) lipoblasts, embedded in a myxoid to collagenous background [23,24]. Given the morphological overlap with atypical lipomatous tumor (ALT)/WDLPS, advances in molecular genetics have undeniably been helpful in a better classification of these atypical adipocytic tumors, as amplification of MDM2 and CDK4, characteristic in ALT/WDLPS is absent in atypical spindle cell/pleomorphic lipomatous tumor. Similar to spindle cell lipoma, these tumors show deletion of 13q, which includes the RB1 gene. They are suggested to represent the more aggressive counterpart of spindle cell lipoma and pleomorphic lipoma, but with the occurrence of atypical spindle cells [25]. However, spindle cell lipomas have a different clinicopathological presentation with well defined margins and predilection for the head and neck region [23].
Anastomosing hemangioma was first described by Montgomery and Epstein and may mimic angiosarcoma. It was initially thought that these tumors were confined to the genitourinary tract, but involvement of other anatomic locations was described soon after [26]. These lesions display one layer of mildly atypical endothelial cells with a hobnail appearance, with a loosely lobulated architecture and are often associated with a medium-sized vessel [26–28]. However, in contrast to angiosarcoma, anastomosing hemangioma are small tumors, the tumors have nonendothelial supporting elements, lobulated architecture without multilayering nor high mitotic activity. An activating hotspot mutation in GNA11, GNAQ, or GNA14 has been reported in up to 93% of the tumors [29–31]. Similar mutations are described in a range of other congenital and small vessel lesions, including hepatic small vessel neoplasm, congenital hemangioma and cherry hemangioma, suggesting a common pathway in the pathogenesis [29,32].
Angiofibroma of soft tissue was firstly described as a distinct benign fibrovascular neoplasm occurring predominantly in the lower extremities of middle-aged adults and growing in close relationship with joints and tendons. This tumor was designated as angiofibroma to reflect the two components of these tumors: relatively uniform, bland spindle cells arranged in a myxoid to more collagenous background and innumerable evenly distributed small thin-walled branching blood vessels [33–36]. In the initial report, cytogenetic analysis already revealed a simple karyotype with a balanced t(5;8) chromosomal translocation [33] and soon here after the specific fusion partners AHRR and NCOA2 were revealed. This translocation is involved in the vast majority of the cases and results in the formation of a chimera transcript that is thought to upregulate the AhR/ARNT signaling pathway. In a minority of cases, alternative fusion partners for NCOA2 are described and in single cases GAB1-ABL1 fusion is involved [35,37,38]. From a diagnostic point of view, genetic confirmation is often not required as the diagnosis can be established based on the distinct morphological features.
Myxoid pleomorphic liposarcoma was first described in a series of liposarcomas in young patients and showed a distinctive and unusual admixture of morphological features characteristic for both myxoid liposarcoma (MLPS) and pleomorphic liposarcoma. These tumors have a predilection for the mediastinum and show a highly aggressive behavior compared with conventional MLPS [39]. Also, from a molecular perspective these tumors are distinct from MLPS and dedifferentiated LPS, as these tumors show a complex karyotype with loss of RB1 while lacking molecular alterations characteristic for MLPS (EWSR1/FUS-DDIT3 fusion) and dedifferentiated LPS (amplification of CDK4 and MDM2) [39–41].
Poorly differentiated chordoma was described as a distinct entity in 2006 [42]. This bone tumor has a predilection for the skull base and cervical spine and occurs most often in children or young adults. It is associated with an aggressive behavior and has a worse prognosis compared with conventional chordoma [42,43]. Morphological features include sheets of relatively small epithelioid cells, often with a rhabdoid appearance, lacking physaliphorous cells [42,43]. The tumor cells express brachyury, S100 and cytokeratin. Besides distinct clinicopathological features, additional molecular data support the classification of poorly differentiated chordoma as an independent subset of chordoma since. This tumor is characterized by deletion of the SMARCB1/INI1. Additional DNA methylation profiling also showed separate clustering of poorly differentiated chordoma from conventional chordoma, in addition to atypical teratoid/rhabdoid tumors, a brain tumor occurring in young children with deletion of SMARCB1/INI-1 as a molecular hallmark [44]. Moreover, SMARCB1 inactivation is believed to lead to loss of inhibition of EZH2, which is targetable using a novel EZH2 inhibitor tazemetostat, as this has been reported in SMARCB1/INI-1 deleted epithelioid sarcoma [43].
CONCLUSIONThe fifth edition of the WHO Classification of Soft Tissue and Bone Tumors includes new entities in which molecular alterations have played an important role in their delineation, either to define or to support their diagnosis. New entities with distinct genetic findings are continuously being described, so it is anticipated that this trend will drive a further refinement in the next edition of the classification [45▪]. For instance, neoplasms defined by GLI1 alterations [46,47] or ‘pseudoendocrine sarcomas’ with recurrent CTNNB1 mutations [48] have recently been reported and further illustrate the dynamic field of molecular diagnostics and their significant role in the recognition of new entities. In addition, further studies are awaited to clarify whether the emerging group of tumors unified by a characteristic molecular rearrangement involving for instance NTRK, but displaying a wide morphological spectrum, an indistinct immunophenotype and a divergent clinical behavior should be considered a single separate entity, solely based on a shared genetic feature or that it includes various entities.
AcknowledgementsNone.
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
REFERENCES AND RECOMMENDED READINGPapers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES 1. Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F. WHO classification of tumours editorial board: soft tissue and bone tumours. 5th ed.Lyon, France: IARC; 2020. 2▪▪. Kallen ME, Hornick JL. The 2020 WHO classification: what's new in soft tissue tumor pathology? Am J Surg Pathol 2021; 45:e1–e23. 3▪▪. Le Loarer F, Baud J, Azmani R, et al. Advances in the classification of round cell sarcomas. Histopathology 2022; 80:33–53. 4. Suurmeijer AJH, Dickson BC, Swanson D, et al. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer 2018; 57:611–621. 5. Antonescu CR. Emerging soft tissue tumors with kinase fusions: an overview of the recent literature with an emphasis on diagnostic criteria. Genes Chromosomes Cancer 2020; 59:437–444. 6. Szuhai K, Ijszenga M, de Jong D, et al. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res 2009; 15:2259–2268. 7. Diaz-Perez JA, Nielsen GP, Antonescu C, et al. EWSR1/FUS-NFATc2 rearranged round cell sarcoma: clinicopathological series of 4 cases and literature review. Hum Pathol 2019; 90:45–53. 8. Ong SLM, Lam SW, van den Akker BEWM, et al. Expanding the spectrum of EWSR1-NFATC2-rearranged benign tumors: a common genomic abnormality in vascular malformation/hemangioma and simple bone cyst. Am J Surg Pathol 2021; 45:1669–1681. 9. Dashti NK, Dickson BC, Zhang L, et al. A unique epithelioid vascular neoplasm of bone characterized by EWSR1/FUS-NFATC1/2 fusions. Genes Chromosomes Cancer 2021; 60:762–771. 10. Pižem J, Šekoranja D, Zupan A, et al. FUS-NFATC2 or EWSR1-NFATC2 fusions are present in a large proportion of simple bone cysts. Am J Surg Pathol 2020; 44:1623–1634. 11. Michal M, Rubin BP, Agaimy A, et al. EWSR1-PATZ1-rearranged sarcoma: a report of nine cases of spindle and round cell neoplasms with predilection for thoracoabdominal soft tissues and frequent expression of neural and skeletal muscle markers. Mod Pathol 2021; 34:770–785. 12. Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol 2017; 41:941–949. 13. Kao YC, Owosho AA, Sung YS, et al. BCOR-CCNB3 fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol 2018; 42:604–615. 14. Specht K, Zhang L, Sung YS, et al. Novel BCOR-MAML3 and ZC3H7B-BCOR gene fusions in undifferentiated small blue round cell sarcomas. Am J Surg Pathol 2016; 40:433–442. 15. Matsuyama A, Shiba E, Umekita Y, et al. Clinicopathologic diversity of undifferentiated sarcoma with BCOR-CCNB3 fusion: analysis of 11 cases with a reappraisal of the utility of immunohistochemistry for BCOR and CCNB3. Am J Surg Pathol 2017; 41:1713–1721. 16. Antonescu CR, Kao YC, Xu B, et al. Undifferentiated round cell sarcoma with BCOR internal tandem duplications (ITD) or YWHAE fusions: a clinicopathologic and molecular study. Mod Pathol 2020; 33:1669–1677. 17. Kao YC, Sung YS, Argani P, et al. NTRK3 overexpression in undifferentiated sarcomas with YWHAE and BCOR genetic alterations. Mod Pathol 2020; 33:1341–1349. 18. Kao YC, Flucke U, Eijkelenboom A, et al. Novel EWSR1-SMAD3 gene fusions in a group of acral fibroblastic spindle cell neoplasms. Am J Surg Pathol 2018; 42:522–528. 19. Michal M, Berry RS, Rubin BP, et al. EWSR1-SMAD3-rearranged fibroblastic tumor: an emerging entity in an increasingly more complex group of fibroblastic/myofibroblastic neoplasms. Am J Surg Pathol 2018; 42:1325–1333. 20. Habeeb O, Korty KE, Azzato EM, et al. EWSR1-SMAD3 rearranged fibroblastic tumor: case series and review. J Cutan Pathol 2021; 48:255–262. 21. Lam SW, Briaire-de Bruijn IH, van Wezel T, et al. NTRK fusions are extremely rare in bone tumours. Histopathology 2021; 79:880–885. 22. Suurmeijer AJ, Dickson BC, Swanson D, et al. The histologic spectrum of soft tissue spindle cell tumors with NTRK3 gene rearrangements. Genes Chromosomes Cancer 2019; 58:739–746. 23. Mariño-Enriquez A, Nascimento AF, Ligon AH, et al. Atypical spindle cell lipomatous tumor: clinicopathologic characterization of 232 cases demonstrating a morphologic spectrum. Am J Surg Pathol 2017; 41:234–244. 24. Dei Tos AP, Mentzel T, Newman PL, Fletcher CD. Spindle cell liposarcoma, a hitherto unrecognized variant of liposarcoma. Analysis of six cases. Am J Surg Pathol 1994; 18:913–921. 25. Creytens D, Mentzel T, Ferdinande L, et al. ‘Atypical’ pleomorphic lipomatous tumor: a clinicopathologic, immunohistochemical and molecular study of 21 cases, emphasizing its relationship to atypical spindle cell lipomatous tumor and suggesting a morphologic spectrum (atypical spindle cell/pleomorphic lipomatous tumor). Am J Surg Pathol 2017; 41:1443–1455. 26. John I, Folpe AL. Anastomosing hemangiomas arising in unusual locations: a clinicopathologic study of 17 soft tissue cases showing a predilection for the paraspinal region. Am J Surg Pathol 2016; 40:1084–1089. 27. Lin J, Bigge J, Ulbright TM, Montgomery E. Anastomosing hemangioma of the liver and gastrointestinal tract: an unusual variant histologically mimicking angiosarcoma. Am J Surg Pathol 2013; 37:1761–1765. 28. Montgomery E, Epstein JI. Anastomosing hemangioma of the genitourinary tract: a lesion mimicking angiosarcoma. Am J Surg Pathol 2009; 33:1364–1369. 29. Liau JY, Tsai JH, Lan J, et al. GNA11 joins GNAQ and GNA14 as a recurrently mutated gene in anastomosing hemangioma. Virchows Arch 2020; 476:475–481. 30. Bean GR, Joseph NM, Folpe AL, et al. Recurrent GNA14 mutations in anastomosing haemangiomas. Histopathology 2018; 73:354–357. 31. Bean GR, Joseph NM, Gill RM, et al. Recurrent GNAQ mutations in anastomosing hemangiomas. Mod Pathol 2017; 30:722–727. 32. Liau JY, Lee JC, Tsai JH, et al. High frequency of GNA14, GNAQ, and GNA11 mutations in cherry hemangioma: a histopathological and molecular study of 85 cases indicating GNA14 as the most commonly mutated gene in vascular neoplasms. Mod Pathol 2019; 32:1657–1665. 33. Mariño-Enríquez A, Fletcher CD. Angiofibroma of soft tissue: clinicopathologic characterization of a distinctive benign fibrovascular neoplasm in a series of 37 cases. Am J Surg Pathol 2012; 36:500–508. 34. Cai Y, Mohseny AB, Karperien M, et al. Inactive Wnt/beta-catenin pathway in conventional high-grade osteosarcoma. J Pathol 2010; 220:24–33. 35. Edgar MA, Lauer SR, Bridge JA, Rizzo M. Soft tissue angiofibroma: report of 2 cases of a recently described tumor. Hum Pathol 2013; 44:438–441. 36. Mindiola-Romero AE, Maloney N, Bridge JA, et al. A concise review of angiofibroma of soft tissue: a rare newly described entity that can be encountered by dermatopathologists. J Cutan Pathol 2020; 47:179–185. 37. Arbajian E, Magnusson L, Mertens F, et al. A novel GTF2I/NCOA2 fusion gene emphasizes the role of NCOA2 in soft tissue angiofibroma development. Genes Chromosomes Cancer 2013; 52:330–331. 38. Jin Y, Möller E, Nord KH, et al. Fusion of the AHRR and NCOA2 genes through a recurrent translocation t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation of aryl hydrocarbon receptor target genes. Genes Chromosomes Cancer 2012; 51:510–520. 39. Alaggio R, Coffin CM, Weiss SW, et al. Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol 2009; 33:645–658. 40. Creytens D, Folpe AL, Koelsche C, et al. Myxoid pleomorphic liposarcoma-a clinicopathologic, immunohistochemical, molecular genetic and epigenetic study of 12 cases, suggesting a possible relationship with conventional pleomorphic liposarcoma. Mod Pathol 2021; 34:2043–2049. 41. Hofvander J, Jo VY, Ghanei I, et al. Comprehensive genetic analysis of a paediatric pleomorphic myxoid liposarcoma reveals near-haploidization and loss of the RB1 gene. Histopathology 2016; 69:141–147. 42. Hoch BL, Nielsen GP, Liebsch NJ, Rosenberg AE. Base of skull chordomas in children and adolescents: a clinicopathologic study of 73 cases. Am J Surg Pathol 2006; 30:811–818. 43. Shih AR, Cote GM, Chebib I, et al. Clinicopathologic characteristics of poorly differentiated chordoma. Mod Pathol 2018; 31:1237–1245. 44. Hasselblatt M, Thomas C, Hovestadt V, et al. Poorly differentiated chordoma with SMARCB1/INI1 loss: a distinct molecular entity with dismal prognosis. Acta Neuropathol 2016; 132:149–151. 45▪. Folpe AL. ‘I Can’t Keep Up!’: an update on advances in soft tissue pathology occurring after the publication of the 2020 World Health Organization classification of soft tissue and bone tumours. Histopathology 2022; 80:54–75. 46. Agaram NP, Zhang L, Sung YS, et al. GLI1-amplifications expand the spectrum of soft tissue neoplasms defined by GLI1 gene fusions. Mod Pathol 2019; 32:1617–1626. 47. Antonescu CR, Agaram NP, Sung YS, et al. A distinct malignant epithelioid neoplasm with GLI1 gene rearrangements, frequent S100 protein expression, and metastatic potential: expanding the spectrum of pathologic entities with ACTB/MALAT1/PTCH1-GLI1 fusions. Am J Surg Pathol 2018; 42:553–560. 48. Papke DJ Jr, Dickson BC, Sholl L, Fletcher CDM. Pseudoendocrine sarcoma: clinicopathologic analysis of 23 cases of a distinctive soft tissue neoplasm with metastatic potential, recurrent CTNNB1 mutations, and a predilection for truncal locations. Am J Surg Pathol 2022; 46:33–43.
留言 (0)