Proteomic analysis reveals that Xbp1s promotes hypoxic pulmonary hypertension through the p‐JNK MAPK pathway
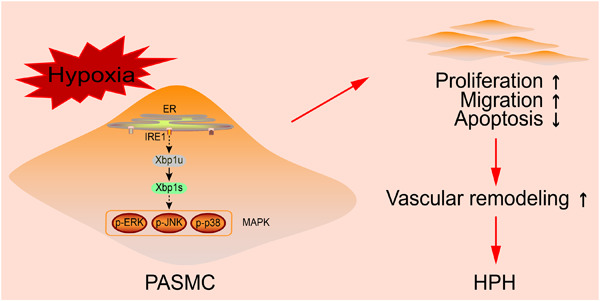
Hypoxic pulmonary hypertension (HPH) is characterized by elevated pulmonary artery resistance and vascular remodeling. Endoplasmic reticulum stress (ERS) is reported to be involved in HPH, but the underlying mechanisms remain uncertain. We found that Xbp1s, a potent transcription factor during ERS, was elevated in hypoxic-cultured rat PASMCs and lung tissues from HPH rats. Our in vitro experiments demonstrated that overexpressing Xbp1s can promote proliferation, cell viability, and migration and inhibit the apoptosis of PASMCs, while silencing Xbp1s led to the opposite. Through data-independent acquisition (DIA) mass spectrometry, we identified extensive proteomic alterations regulated by hypoxia and Xbp1s. Further validation revealed that p-JNK, rather than p-ERK or p-p38, was the downstream effector of Xbp1s. p-JNK inhibition reversed the biological effects of Xbp1s overexpression in vitro. In the animal HPH model, rats were randomly assigned to five groups: normoxia, hypoxia, hypoxia+AAV-CTL (control), hypoxia+AAV-Xbp1s (prevention), and hypoxia+AAV-Xbp1s (therapy). Adeno-associated virus (AAV) serotype 1-mediated Xbp1s knockdown in the prevention and therapy groups significantly reduced right ventricular systolic pressure, total pulmonary resistance, right ventricular hypertrophy, and the medial wall thickness of muscularized distal pulmonary arterioles; AAV-Xbp1s also decreased proliferating cell nuclear antigen expression and increased apoptosis in pulmonary arterioles. Collectively, our findings demonstrated that the Xbp1s-p-JNK pathway is important in hypoxic vascular remodeling and that targeting this pathway could be an effective strategy to prevent and alleviate HPH development.




留言 (0)