Figure S1. Estimating the maternal-fetal genotype of a polymorphic site using its allelic read counts. R1, R2 and R3: allelic read counts in descending order; α: background noise threshold. A, B and C are distinct alleles for each polymorphic site, and the portion before the vertical bar denotes the maternal genotype and the part after the vertical bar denotes the fetal genotype. FC: estimated reads count amplified from fetal genetic materials (Fetal Reads); TC: total reads count amplified from both maternal and fetal genetic materials (Total Reads).
Figure S2. Fetal fraction estimation using allelic read counts or whole genome sequencing. Samples from the insertion/deletion polymorphism dataset were analyzed. Chromosomes X and Y were divided into non-overlapping 50 kb bins, and reads in each bin were counted for each sample. Median bin read counts for chromosomes X and Y of each sample were plotted (A). (B-D) Fetal fractions were estimated for each sample by both the allelic read counts and the WGS methods, and their relationship was plotted (red line: regression line y ~ x). (B) All samples. (C) Excluding samples with the median bin read count of chromosome X < 100. (D) Excluding samples with both median (ChrX) < 100 and median (ChrY) < 4. Same as Figure 1E, included here for comparison purposes.
Figure S3. Estimating fetal fractions for samples in the replication dataset. (A) Samples from the replication dataset were analyzed and fetal fraction of each sample was estimated using the allelic read counts method. (B) Replicated samples were grouped and mean fetal fraction (±SD) was plotted for each group.
Figure S4. Estimation accuracy for fetal fractions of simulated samples. Samples were simulated for different reads coverages and different fetal fractions. One hundred samples were simulated for each coverage and 100 polymorphic sites were simulated for each sample. Expected fetal fraction: the fetal fraction value used for simulating sequencing reads. Estimated fetal fraction: the fetal fraction estimated using allelic reads counts of each sample. C: coverage.
Figure S5. Relative allelic counts plot. One hundred polymorphic sites on a disomy-disomy chromosome were simulated for each sample. For each polymorphic site in a sample, reads counts of different alleles were sorted in descending order and labeled as R1, R2 and R3, respectively. One representative plot was shown for each fetal fraction. f: fetal fraction. Relative R1 Count = R1/(R1 + R2 + R3) and Relative R2 Count = R2/(R1 + R2 + R3).
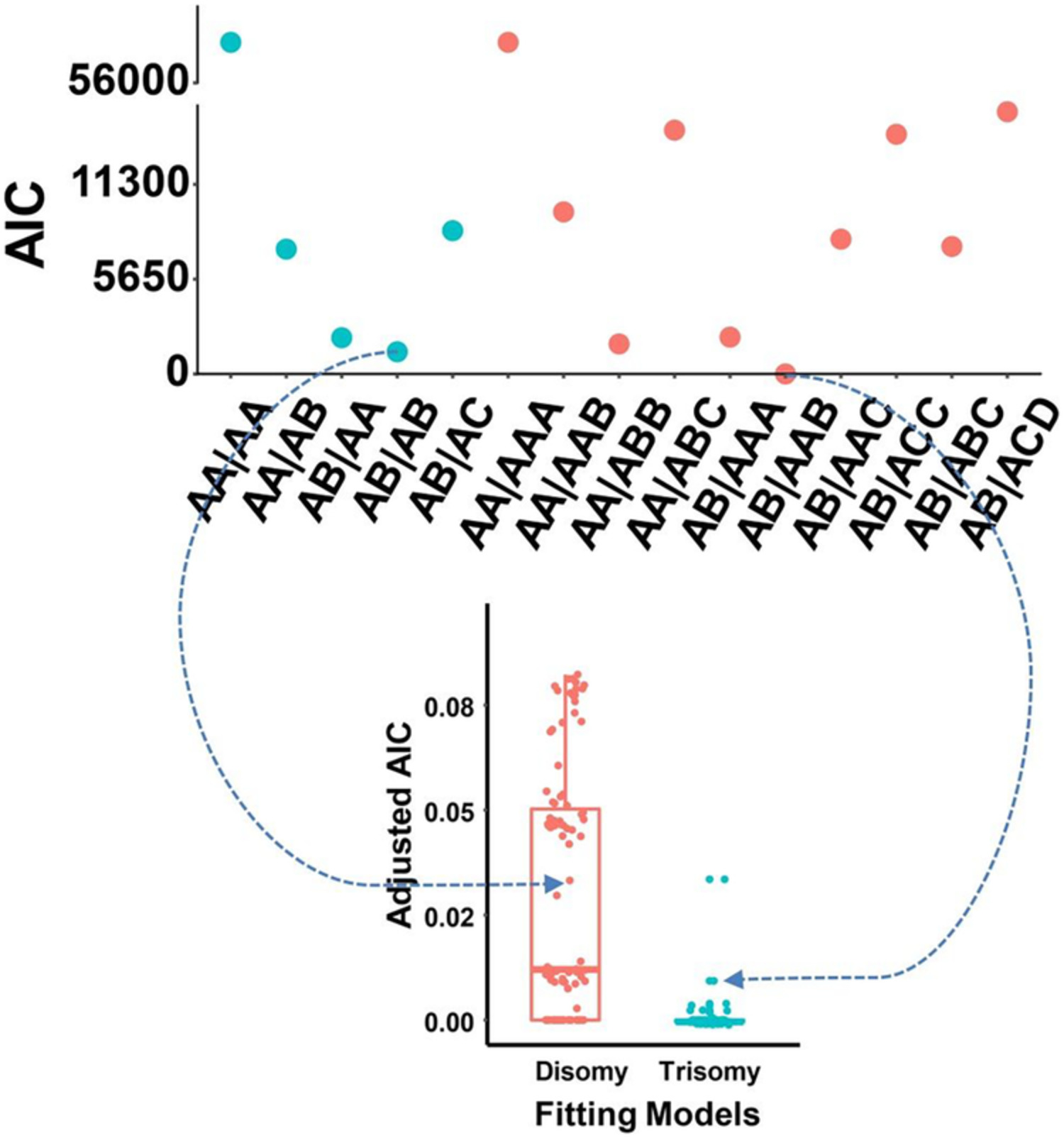
Figure S6. Influences of total reads count on the statistics of ΔAIC and adjusted ΔAIC. Samples from the replication dataset were analyzed and the genotype of each polymorphic site was estimated followed by the calculation ΔAIC and adjusted ΔAIC. ΔAIC = absolute AIC differences between the two best fitted models. Adjusted ΔAIC = ΔAIC/TotalCount/FetalFraction. The ΔAIC (A) and adjusted ΔAIC (B) were plotted against the total reads count of each polymorphic site.
Figure S7. Estimation accuracy for genotypes of simulated samples. Samples were simulated for different reads coverages and different fetal fractions. One hundred samples were simulated for each coverage and 100 polymorphic sites were simulated for each sample. Estimation accuracy: the ratio of the number of correctly estimated genotypes to the total number of all polymorphic sites in each group. C: coverage.
Figure S8. Estimation accuracy for genotypes of the replication dataset. Polymorphic sites of replicated samples were analyzed individually (1) or in a group (2-4), whereas genotype of each polymorphic site was estimated using 1, 2, 3 or 4 replicated samples, respectively. Estimation accuracy: the ratio of the number of correctly estimated genotypes to the total number of polymorphic sites in a group.
Figure S9. Detecting pathogenic fetal variants for the wilson validation dataset. In the 5% fetal fraction group of the wilson validation dataset, 8 sample replicates were prepared using maternal and paternal plasma samples mimicking homozygous-heterozygous maternal-fetal genotypes. Relative allelic counts plot (left) and the allelic goodness of fit test (right) results were plotted in a group for all replicated samples. Since allele A was wildtype, the allelic clusters (left) and the best fit model (AA|AB) indicated that the mother was homozygous wildtype and the fetus was heterozygous for the pathogenic variant.
Figure S10. Detecting pathogenic fetal variants for the wilson validation dataset. In the 10% fetal fraction group of the wilson validation dataset, 6 sample replicates were prepared using maternal and paternal plasma samples mimicking homozygous-heterozygous maternal-fetal genotypes. Relative allelic counts plot (left) and the allelic goodness of fit test (right) results were plotted in a group for all replicated samples. Since allele A was wildtype, the allelic clusters (left) and the best fit model (AA|AB) indicated that the mother was homozygous wildtype and the fetus was heterozygous for the pathogenic variant.
Figure S11. Detecting pathogenic fetal variants for the wilson validation dataset. In the maternal heterozygous group of the wilson validation dataset, 11 maternal plasma samples were prepared and each had a heterozygous fetus for the target pathogenic variant. As fetal fraction was different for each sample and the estimations of fetal fractions were reported in only 5 of the 11 samples, all samples were analyzed in a group assuming each one had the average fetal fraction of the 5 reported values mimicking replicate samples of heterozygous-heterozygous maternal-fetal genotypes. Relative allelic counts plot (A) and the allelic goodness of fit test (B, C) results were plotted. Since allele A was wildtype, the allelic clusters (A) and the best fit model (AB|AB) indicated that both the mothers and the fetuses were heterozygous for the pathogenic variant.
Figure S12. Analyses of genotype estimation results for monogenic variant screening datasets. Maternal-fetal genotype of each pathogenic variant hot site for the hbb dataset (A), arnshl dataset (B) and cfbest dataset (C) was estimated. The Adjusted ∆AIC values were plotted for sites estimated correctly (concordant) and incorrectly (discordant) for each dataset.
Table S1. Relative Allelic Counts for a Polymorphic Site on Reference Chromosomes
Table S2. Relative Allelic Count for Disomy-Disomy Model
Table S3. Relative Allelic Count for Disomy-Monosomy Model
Table S4. Relative Allelic Count for Disomy-Trisomy Model
Table S5. Detection Accuracy of Chromosomal Aneuploidies for Simulated Samples
Table S6. Genotype Estimation for a Two-allele Site
Table S7. Genotype Estimation for a Site with More Than Two Alleles
Table S8. Genotype Estimation for Fetal Wilson Diseases Using Plasma Samples
Table S9. Genotype Estimation for the hbb Dataset Using Maternal Plasma Samples
Table S10. Genotype Estimation for the arnshl Dataset Using Maternal Plasma Samples
Table S11. Genotype Estimation for the cfbest Dataset Using Maternal Plasma Samples
Table S12. Genotype Estimation Results for the wilson Validation Dataset Using Plasma Mixtures








留言 (0)