
記住我

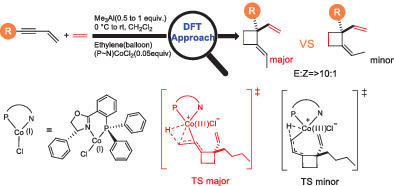

There has been tremendous advancement in machine learning (ML) applications in computational chemistry, particularly in neural network potentials (NNP). NNPs can approximate potential energy surface (PES) as a high dimensional function by learning from existing reference data, thereby circumventing the need to solve the electronic Schrödinger equation explicitly. As a result, ML accelerates chemical space exploration and property prediction compared to quantum mechanical methods. Novel ML methods have the potential to provide efficient means for predicting the properties of molecules. However, this potential has been limited by the lack of standard comparative evaluations. In this work, we compare four selected models, that is, ANI, PhysNet, SchNet, and BAND-NN, developed to represent the PES of small organic molecules. We evaluate these models for their accuracy and transferability on two different test sets (i) Small organic molecules of up to eight-heavy atoms on which ANI and SchNet achieve root mean square error (RMSE) of 0.55 and 0.60 kcal/mol, respectively. (ii) On random selection of molecules from the GDB-11 database with 10-heavy atoms, ANI achieves RMSE of 1.17 kcal/mol and SchNet achieves RMSE of 1.89 kcal/mol. We examine their ability to produce smooth meaningful surface by performing PES scans for bond stretch, angle bend, and dihedral rotations on relatively large molecules to assess their possible application in molecular dynamics simulations. We also evaluate their performance for yielding minimum energy structures via geometry optimization using various minimization algorithms. All these models were also able to accurately differentiate different isomers of the same empirical formula  . ANI and PhysNet achieve an RMSE of 0.29 and 0.52 kcal/mol, respectively, on
. ANI and PhysNet achieve an RMSE of 0.29 and 0.52 kcal/mol, respectively, on  isomers.
isomers.
留言 (0)