
記住我
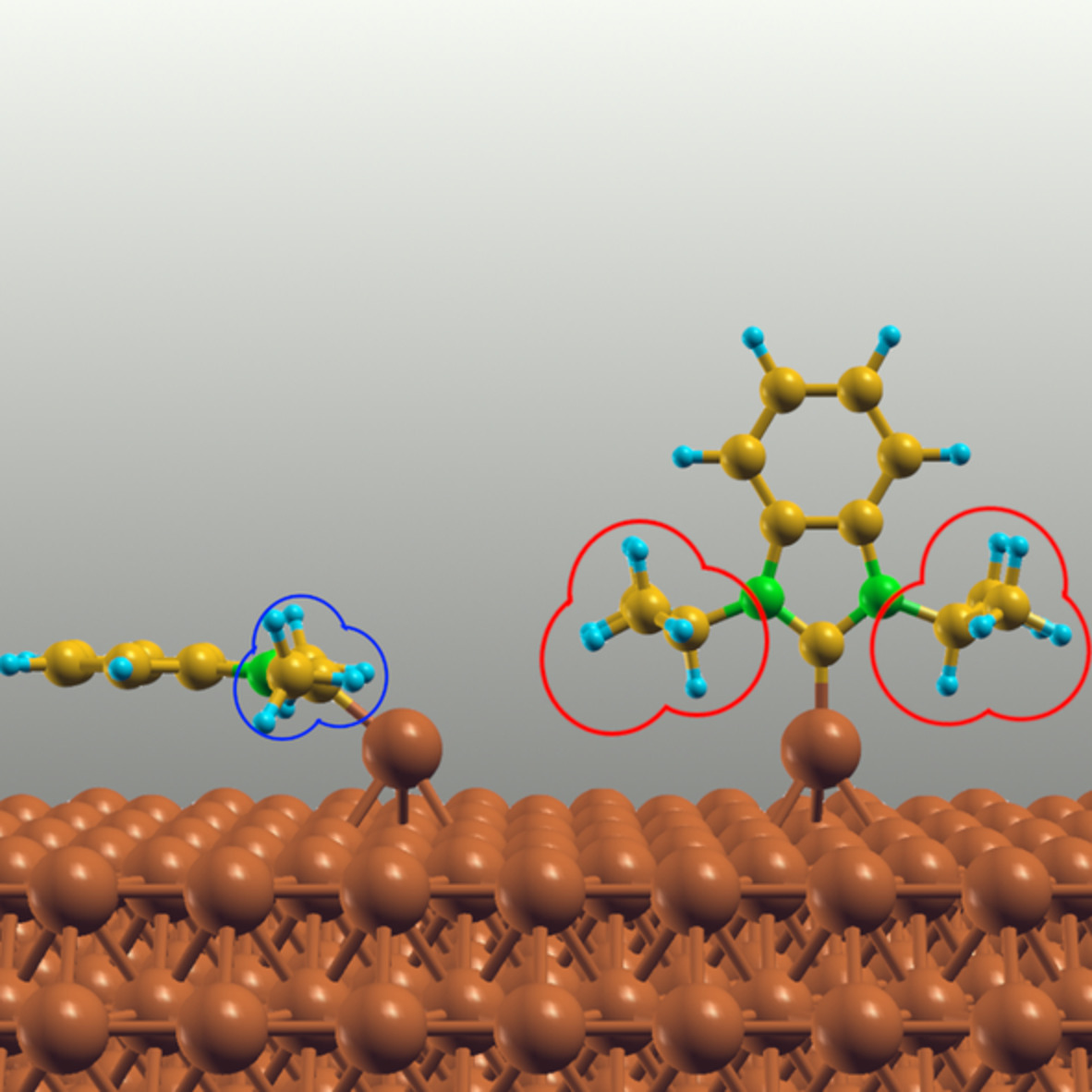


We start our DFT calculations by carefully determining the most stable structures of the three NHCs on pristine Au(111) and Cu(111) surfaces. Since the registries of the molecules depend on the flexible orientation of the side groups and the orientation of the molecule itself and provide a flat energy landscape, a large variety of plausible starting configurations have to be used and fully relaxed to identify the ground state geometries and possible intermediate states. This includes different spatial geometries (flat-laying, perpendicular and tilted), different rotational degrees of freedom and different surface-related registries (top, bridge, fcc and hcp hollows) of the adsorbed NHC molecule (see Figure S1 and Tables SIII and SIV for details).
In agreement with previous works,9, 16, 30 the most stable structures of the three NHCs on ideal surfaces are characterized by a formation of a single covalent bond between the NHC's carbene carbon atom (see Figure 1) and one atom from the metal surfaces (see Figure 2, Figure S3, and the charge density difference plots in Figure S6). Such geometries are denoted as surface-bound species in the following. Table 1 compares the characteristic structural parameters, the adsorption energies and the contributions of the dispersion interactions to the adsorption energies for the three surface-bound NHCs in their most stable states.

Side-views of the most stable adsorption configuration of (exemplarily shown) NHC1 on the ideal Au(111). The carbene carbon atom is covalently bound to one of the substrate atoms (bond length d1). The tilting angle (θ) is defined between the N-heterocyclic carbenes (NHC) ring plane and the surface normal. The surface atom is lifted upwards by d2 (to better show this parameter, the surface atoms are shown in a stick display-mode)
TABLE 1. A comparison of most stable structures of NHC1, NHC2, and NHC3 on Cu(111) and Au(111). The contributions of the van der Waals (vdW) interactions (EvdW) to the adsorption energy (Eads) are also given. The adsorption geometry parameters (d1, d2, and θ) have been defined in Figure Configuration Eads (eV) EvdW (eV) (% Eads) d1 (Å) d2 (Å) θ (degree) Cu(111) Au(111) Cu(111) Au(111) Cu(111) Au(111) Cu(111) Au(111) Cu(111) Au(111) Cu(111) Au(111) NHC1 upright upright −1.96 −1.89 −1.03 (53%) −0.85 (45%) 2.05 2.14 0.43 0.36 3 7 NHC2 upright upright −2.28 −2.16 −1.32 (58%) −1.12 (52%) 2.05 2.15 0.52 0.44 7 6 NHC3 upright upright −2.58 −2.43 −1.62 (63%) −1.40 (58%) 2.05 2.16 0.60 0.48 0 0The calculated d1 values for the adsorption heights are in very good agreement with those reported for other NHC on Au(111),8, 14, 15, 25, 29, 30, 32 and slightly longer than that reported in Reference [31] (1.97 Å) for an NHC on Cu(111). However, this value is apparently shorter than that reported in the recent work of Deng et al.32 (2.21 Å) and might be related to the laterally smaller supercells used in Reference [31]. In any case, for a given substrate the contribution of the covalent interactions (−0.85 eV on Cu(111) [−1.05 eV on Au(111)]), and by this the bond length (d1 = 2.05 Å) (~2.15 Å) are almost the same for the three NHCs. The most striking effect onto structure is thus given by the influence of the substrate, as the bond lengths on the more reactive Cu(111) surface are shorter by about 5% compared to adsorption on Au(111) surfaces. Consequently, the adsorption energies for the three NHCs are slightly higher on Cu(111) compared to Au(111). A similar trend was observed for NHC3 in Reference [12]. However, an opposite tendency was reported by Deng et al.32 for NHC1, where surprisingly high adsorption energies on Au(111) were obtained.
Our calculations clearly show, however, that the adsorption energies are increasing with the size of the side groups. Apparently, this is related to the increased contribution of the dispersion interactions of 53% (45%), 58% (52%) and 63% (58%) for NHC1, NHC2, and NHC3 on Cu(111) (Au(111)), respectively. The vertical shift of the metal atom on which the molecule anchors (d2) shows the same tendency as well, further highlighting the crucial role of the dispersion interactions to determine adequate molecular geometries and reliable adsorption energies, also in case of covalently bound species.
It is important to note that our calculations found no indications for any flat-laying geometries. Irrespective of the start structure, the geometries of all studied molecules have been finally converged to upright configurations (with θ < 10°; see Table 1). Vertical geometries (θ = 0°) can be achieved within a few meV. In contrast, forcing the molecules to adopt flat-laying geometries would require energies in the order of 1 eV (see Figure S2 for details). This is true not only on Au(111),14, 30 but also on Cu(111). In both cases, flat-laying adsorption geometries can be safely ruled out on pristine metallic substrates and thus cannot account for the different binding modes reported by Larrea et al.12 for these NHCs.
3.2 Adsorption at Cu/Au adatomsAdsorption at step edges or at omnipresent adatoms might facilitate a flat adsorption scheme (see Figure 3). Such scenarios were previously reported for isonitriles,37 thiols38 and NHCs on Au(111).9 According to Wang et al.9 the energy cost for the extraction of an Au atom (to form an adatom), is reduced by one order of magnitude (to the 100 meV regime) if small-sized NHC are attached. This premise has been verified by STM height measurements,9 which confirmed the adsorption of NHCs at adatoms on the Au(111) surface. On Cu (111), extracting Cu atoms from steps and their incorporation in the molecular assembly is reported to be a thermally activated process.39
 (A) Side-view of, for example, an Au adatom residing on the fcc hollow site of planar surface. Its height assuming no molecular adsorption is considered as a reference (d0). (B,C) side-views of the most stable structures of a N-heterocyclic carbenes (NHC) (e.g., NHC2) adsorbed on an adatom (ballbot species) in an upright/a flat-laying geometry. The vertical upwards shift of the adatom after the NHC adsorption is denoted as d3. (D) Is like (C) but in a top-view. The azimuthal orientation of the molecular symmetry axis (SA) with respect to the direction of the surfaces is denoted as (ϕ). The adsorption parameters of NHC1, NHC2, and NHC3 are given in Table 2 and 3 for Cu(111) and Au(111), respectively
(A) Side-view of, for example, an Au adatom residing on the fcc hollow site of planar surface. Its height assuming no molecular adsorption is considered as a reference (d0). (B,C) side-views of the most stable structures of a N-heterocyclic carbenes (NHC) (e.g., NHC2) adsorbed on an adatom (ballbot species) in an upright/a flat-laying geometry. The vertical upwards shift of the adatom after the NHC adsorption is denoted as d3. (D) Is like (C) but in a top-view. The azimuthal orientation of the molecular symmetry axis (SA) with respect to the direction of the surfaces is denoted as (ϕ). The adsorption parameters of NHC1, NHC2, and NHC3 are given in Table 2 and 3 for Cu(111) and Au(111), respectively
Adatoms are also reported to play a key role for the formation of NHC self-assembled monolayers.40 The presence of adatoms explains the high mobility of NHCs on Au(111) surface despite strong NHCAu bond formation. As suggested by Wang et al.9 for 1,3-dimethylimidazol-2-ylidene (IMe) molecule and by Larrea et al.12 for NHC1, NHC2, and NHC3, the formation of NHC–adatom complexes enables the so-called ‘ballbot’-type motion of these complexes. It offers low barriers to diffuse, much lower than surface-bound molecules and even lower than the adatom alone, thereby promoting the molecular self-assembly. As a simple test for this premise, we calculate the migration barriers of NHC3 along the <110> direction of Au(111). Barriers of about 0.9 eV for single surface-bound NHC3 are lowered to about 0.2 eV for the NHC3–Au ‘ballbot’ complex (see Figure S4).
3.2.1 N-heterocyclic carbene ballbot species on Cu(111) and Au(111) surfacesIn the following, we show that this NHC ballbot species, that is, adsorption on surface (M) adatom on M(111) surface, M@M(111), is actually able to explain the different adsorption modes experimentally observed.12 In agreement with Wang et al.,9 single Au [Cu] adatoms are found to be most stable when they occupy fcc hollow sites of the surface at a distance of d0 = 2.00 Å (1.86 Å). In order to decorate these adatoms with NHCs, we tested a wide variety of plausible start geometries of NHC1, NHC2, and NHC3 in various ballbot configurations. Notably, in contrast to the surface-bound species, we found for each species stable upright (standard ballbot) and flat-laying structures on both metal surfaces (see Figure 3).
Table 2 presents the structural geometries and the adsorption energies of the most stable structures of the three NHCs on Cu(111) in the upright and flat-laying binding modes (see also Figure S5). Compared to the surface-bound species, NHC ballbot species in the upright geometries are further stabilized by about 0.85, 0.80, and 0.67 eV for NHC1, NHC2, and NHC3, respectively (Figure 5A). This can be related to the fact that the metal s- and d-orbitals are more accessible in the ballbot species, enabling a stronger donor–acceptor bond.14 Thereby, the length of the formed CCu bond (about 1.93 Å) is apparently shorter than that for the surface-bound species (2.05 Å). This tendency is also reflected in the far smaller upwards shift of the adatom (d3) compared to that (d2) shown in Table 1 for the same NHCs in surface-bound species.
TABLE 2. Adsorption parameters of most stable structures of ballbot species of NHC1, NHC2, and NHC3 on Cu(111). For definition of d1, d3, the angles θ, see text and the caption of Figure . Compared to the upright structures, the description of the flat-laying geometries requires an extra parameter. It is the azimuthal orientation of the molecular symmetry axis (SA) with respect to the surface planes (ϕ) and its value is dependent on the investigated NHC Eads (eV) EvdW (eV) (% Eads) d1 (Å) d3 (Å) θ (degree) ϕ (degree) Upright Flat Upright Flat Upright Flat Upright Flat Upright Flat Upright Flat NHC1 −2.80 −3.12 −0.65 (23%) −1.51 (49%) 1.94 1.96 0.06 0.13 0 85 – 11 NHC2 −3.09 −3.29 −0.92 (30%) −1.65 (50%) 1.93 1.96 0.03 0.13 0 85 – 9 NHC3 −3.25 −3.08 −1.05 (33%) −1.93 (63%) 1.92 1.96 0 0.14 0 86 – 30In parallel, the flat-laying ballbot geometries become possible and even most stable for NHC1 and NHC2 (see also Figure 5A). In fact, further 0.32 and 0.20 eV, respectively, are gained in comparison with upright adsorption on Cu@Cu(111). However, in case of NHC3, the upright adsorption remains favored by 0.17 eV. These results are fully in line with the measurements of Larrea et al.12 and can be understood in the light of a subtle balance between the covalent CCu bond on the one hand and the van der Waals (vdW) energy interactions between the molecule and planar surface on the other hand. Going from upright to flat-laying geometries yields a twist in the CuC bonds accompanied with energy-loss of 0.54, 0.53 and 1.05 eV, for NHC1, NHC2, and NHC3, respectively. This is reflected in the increased length of the CCu bond (d1) and upwards shifts Cu adatom (d3) in the latter. In parallel, it gives rise to higher vdW interactions for the respective species by 0.86, 0.73, and 0.88 eV. Without adatoms (i.e. for surface-bound species), the molecule (or at least parts of it) would be simply too close to the surface atoms, so that flat-laying geometries are not possible from steric reasons. It is thus intuitively clear that the upright configuration provides the mobile version of any NHC ballbot; it will become dominating at elevated temperatures, where thermal fluctuations of the side groups decrease the influence of vdW interactions.
The results drawn for Cu(111) can be more or less transferred to the Au(111) surface, see Table 3. Compared to the surface-bound species, NHC ballbot species in the upright geometries are more stable by about 1 eV, in agreement with previous studies.14, 16, 29, 30 The length of the formed CAu bond (2.03 Å) is again reduced (2.15 Å in the surface-bound species). The calculated length of d1 is in a very good agreement with values presented in References [13, 14, 16, 23, 29]. A comparison between upright and flat-laying geometries indicates that the length of d1 is longer in the latter by about 2%. The tilting angles of the three NHCs on Au(111) show, however, a different tendency compared to Cu(111): The flat-laying molecules are completely parallel to the surface θ ~ 90°, while the upright configurations exhibit different tilting angles of 6°, 18°, and 35° for NHC1, NHC2, and NHC3, respectively. Such unexpected geometries indicate that the size of the side groups plays a more decisive role. The molecular geometries are the result of complicated molecular-surface interactions and their detailed balance with respect to the size of the molecular side groups.
In addition, in this case the calculations are in agreement with the experimental observations.12 The adsorption energies of NHC1 and NHC2 [NHC3] in the flat-laying geometries are more stable (less stable) than the upright ones by 0.41 and 0.20 (0.09) eV (see Figure 5B). Notably, a weak dependence of the adsorption energies for NHC3 on the tilting angle was previously reported.14 In agreement with their near-edge X-ray absorption fine-structure spectroscopy (NEXAFS) measurements for NHC3 on Au(111), the calculations show that NHC3 adopt an intermediate tilting angle of θ = 35° nicely reflecting the experimentally derived value of about 40°.
3.2.2 Bis(N-heterocyclic carbene) complexes on Cu(111) and Au(111)Our calculations in the previous section demonstrate that single NHC ballbot species are energetically more favorable than surface-bound species. Another conceivable adsorption scenario is the formation of flat-laying bis(NHC) species, where both NHC molecules are bound to the same surface atom (NHC–adatom–NHC, see Figure 4). For different small-sized NHCs (IMe and NHC1), such species have been previously reported experimentally on Au(111) as well as on Cu(111) surfaces, see for example, References [9, 10, 12, 13]. There are some indications that the formation of such complexes is promoted by thermal activation or by intermolecular interactions.12, 14
 Side views of the most stable adsorption configuration of bis(N-heterocyclic carbenes (NHC)) complexes on Cu(111) and Au(111) surfaces: (A) (NHC1)2Cu and (B) (NHC3)2Au. The height of the adatom without molecular adsorption is considered as a reference (d0). The definitions of d1, d3, and θ were presented in Figure 3
Side views of the most stable adsorption configuration of bis(N-heterocyclic carbenes (NHC)) complexes on Cu(111) and Au(111) surfaces: (A) (NHC1)2Cu and (B) (NHC3)2Au. The height of the adatom without molecular adsorption is considered as a reference (d0). The definitions of d1, d3, and θ were presented in Figure 3
Table 4 summarizes the calculated adsorption geometries and energies for the most stable bis(NHC) structures for NH1, NHC2, and NHC3 on both substrates. In all cases, the adsorption energies per molecules are comparable to those obtained for the ballbot species (cf. Tables 2 and 4). They are, thus, again much larger than those obtained for surface-bound NHC (see also Figure 5 for an overview): bis(NHC) dimer formation is energetically more favorable by 0.8 eV (NHC3) to 1.3 eV (NHC1, NHC2) on Au(111) and 0.5–1 eV on Cu(111), respectively. This indicates that the formation of ballbot species, in particular in flat-laying geometry, provides a highly efficient precursor for the formation of bis(NHC) species, independent on the size of the molecular side group. On Au(111), the adsorption energy per molecule for the (NHC)2Au dimers are even slightly higher (by 10–80 meV) than the adsorption energies of the single NHC flat-lying ballbot species. In other words, the (NHC)2Au dimers are highly probable and expected to be formed even prior to a separate ballbot configuration at already present, but still ‘unoccupied’ adatoms.
TABLE 3. Adsorption parameters of most stable structures of ballbot species of NHC1, NHC2, and NHC3 on Au(111). The definition of d1, d3, the angles θ and ϕ, were presented in Table Eads (eV) EvdW (eV) (% Eads) d1 (Å) d3 (Å) θ (degree) ϕ (degree) Upright Flat Upright Flat Upright Flat Upright Flat Upright Flat Upright Flat NHC1 −2.83 −3.24 −0.41 (15%) −1.28 (40%) 2.03 2.07 0.13 0.10 6 89 – 30 NHC2 −3.15 −3.35 −0.70 (22%) −1.42 (43%) 2.03 2.07 0.11 0.14 18 89 – 30 NHC3 −3.32 −3.23 −0.95 (29%) −1.62 (50%) 2.04 2.07 0.10 0.15 35 89 – 30 TABLE 4. Formation of (NHC)2Cu [(NHC)2Au] complexes from NHC1, NHC2, and NHC3 on Cu(111) (Au(111)) surfaces. The contributions of the vdW interactions (EvdW) to the adsorption energy per molecule (Eads) are also given. The adsorption geometry parameters (d1, d3, and θ) have been defined in Figure and Figure Eads (eV) EvdW (eV) (% Eads) d1 (Å) d3 (Å) θ (degree) Cu(111) Au(111) Cu(111) Au(111) Cu(111) Au(111) Cu(111) Au(111) Cu(111) Au(111) NHC1 −2.96 −3.25 −1.59 (54%) −1.32 (41%) 1.97 2.05 0.49 1.37 85 90 NHC2 −3.22 −3.43 −1.80 (56%) −1.50 (44%) 1.96 2.04 0.45 1.22 85 90 NHC3 −3.06 −3.25 −2.15 (70%) −1.70 (53%) 1.99 2.06 0.56 1.39 85 90
The plots showing comparison of adsorption energies (eV, per molecule) for different configurations considered in this work with respect to NHC1, NHC2, and NHC3 molecules adsorbed on (A) Cu(111) and (B) Au(111)
On Cu (111), the situation is more complex. The adsorption energies per molecule for (NHC)2Cu are slightly smaller than for the respective most stable ballbot species (cf. Figure 5A), suggesting that on Cu(111) the formation of dimers requires high NHC coverage. At least at low temperatures, all available Cu adatoms should be first decorated with a single NHC, before the formation of bis(NHC)Cu complexes sets in. We have to note, however, that further alternative formation scenarios exist and that dimer formation could be promoted kinetically at higher temperatures when the bridging Cu adatom is lifted and the potential (NHC)2Cu dimers become mobile. An alternative formation scenario for bis(NHC) dimers has been indeed suggested in Reference [13] and is confirmed by our present calculations: Starting from NHCs binding to the same surface atom, structural relaxation always converges to geometries, where the metallic atom is pulled out of the surface, leaving a vacancy behind (see Supplementary movie). In case of Au(111), the surface atom is completely detached from the surface layer, resulting barrier-free in an almost planar, physisorbed NHC–Au–NHC dimer configuration, see also Figure 4B, where the central Au atom lay in plane with the two NHC molecules, in agreement with NEXAFS measurements reported in Reference [14].
On Cu(111) our calculations find that a similar planar configuration is less stable, by about 0.2 eV higher in energy than the most stable structure, where the Cu atom within the (NHC)2Cu dimer is still covalently bound to two surface atoms (bridging position, see also Figure 4A). The central Cu adatom resides below the two NHCs, hindering them to adsorb completely parallel to the surface (θ = 85°). Such result is in line with STM measurements reported in Reference [10], where the central Cu atom is observed to appear less bright than the two NHC molecules. This again contributes in rationalizing the different, strongly surface depending binding modes observed by Larrea et al.12
留言 (0)