
記住我






The neuropathological spectrum of fronto-temporal lobar degeneration (FTLD) phenotypes has expanded during the last decade. FTLD with tau molecular signature (FTLD-Tau) comprises classic pathologies such as Pick's disease (3-repeat tauopathy) and progressive supranuclear palsy, corticobasal degeneration, argyrophilic grain disease and globular glial tauopathies as the main representatives of the 4-repeat tauopathies. All show neuronal and particularly glial (astroglial and oligodendroglial) tau accumulation.1 Age-related tau astrogliopathy (ARTAG) is a recent consensus term for age-related astroglial tau pathology, which is not necessarily associated with cognitive decline.
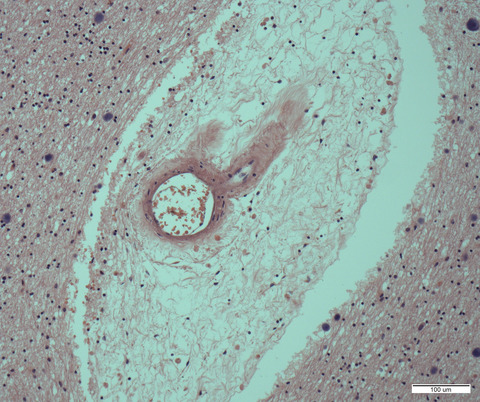
In 2015, Ferrer et al. described an astroglial predominant tauopathy manifesting as familial behavioural FTD.2 Since then, we have identified two additional unrelated individuals with the same neuropathological features in the Barcelona Brain Bank. This relatively novel tauopathy is characterized by prominent p62 (clone 3/p62 lck ligand, BD Biosciences, San Jose, USA; 1:500) and pTau (clone AT8, Thermo Scientific, Rockford, USA; 1:1000) immunoreactivity mainly in astrocytes, particularly of the glia limitans of the perivascular, subpial regions and subependymal regions and throughout the grey and white matter (Figure 1). This astroglial pathology is particularly striking in the cerebellar cortex (Figure 1D,H,M-O) where astrocytic processes of the Bergmann glia surround Purkinje cells and extend into the molecular layer. This pattern and regional involvement seem to be a quite unique identifying feature of this disease. AT8 positive astrocytes show a spectrum of morphologies that resemble thorn-shaped astrocytes (Figure 1Q), tufted astrocytes (Figure 1K,R) and rarely also astrocytic plaques (Figure 1S).

Cortical and subcortical pathology of the new cases identified at the Barcelona Brain Bank. A-D, Overview of cortical (A, HE) and cerebellar (C, HE) extensive tau astrogliopathy (B, D, tau immunohistochemistry). E-J, I, AT8 immunohistochemistry shows prominent tau pathology in cortical (E), limbic (F) and subcortical areas (G, thalamus) including the cerebellar cortex (H). The tau astrogliopathy is the most striking feature. It involves most astrocytes of the grey and white matter and particularly the perivascular astrocytes (I; Gallyas; J: AT8) and subpial astrocytes (Q). K, L, R, S, T, The morphology of astrocytic pathology varies and combines tufted-like (K, L, R), thorn-shaped like, bizarre morphologies and very rarely astrocytic-plaque-like structures (S) These astroglial inclusions are immunoreactive for p62 (K) and predominantly for 4R tau isoforms (L, upper panel), but also for 3R isoforms (L, lower panel). They are also immunoreactive for phosphorylated TDP-43 protein (T, various morphologies). M-P, The cerebellar involvement is very characteristic, particularly of the Bergmann glia (molecular layer). Astrocytes are immunoreactive for Tau-AT8 (M), p62 (N) and phosphorylated TDP-43 (O). In contrast, full-length TDP-43 shows only very isolated positive cells (P, arrow). U-X, In the dentate gyrus there is moderate tau positive astroglial pathology around granule cells associated with a moderate amount of pTDP-43 neuronal cytoplasmic inclusions. Y-Z1, In cortical areas, HE-stained sections reveal astrocytes with enlarged and pleomorphic nuclei (HE) that show AT8 (Y1) and p-TDP-43 (Z) positive inclusions. Full-length TDP-43 shows large negative astrocytic nuclei, without cytoplasmic immunoreactivity (Z1, upper panel, arrows). pTDP-43 reveals astrocytes with diffuse granular cytoplasmic staining (Z1 lower panel). Scale bars: 10 μm for R, S, Y, Y1, Z, Z1; 20 μm for I, J, K, L, M, N, O, P, T, U, V, W, X; 50 μm for E, F, G, Q; 100 μm for A, H; 200 μm for B; 300 μm for C, D
In the cerebral cortex and white matter, the astrocytes appear highly activated, and show enlarged and pleomorphic nuclei on HE-stained sections (Figure 1Y). Neuronal involvement is less prominent and is characterized by large tangles in basal ganglia, N. basalis Meynert, thalamic and brainstem neurons, similar to PSP. This tauopathy is dominated by 4-repeat tau isoforms (clone 1E1/A6, Millipore, Temecula; 1:50) (Figure 1L, upper panel), but not exclusively, as a 3R tau isoform component (clone 8E6/C11, Millipore; 1:5000) of fibrillar neuronal and glial inclusions is also present (Figure 1L, lower panel). In case 2 there was also segmental hippocampal sclerosis that involved the CA1 sector and subiculum. This was associated with abundant TDP-43-positive (clone 2E2-D3, Abnova, Taipei, Taiwan; 1:500) thin neurites, which were also identified in a high amount in case 1.
The cases reported by Ferrer also presented TDP-43 pathology to some extent but was not described in detail. Those cases were originally stained with the full-length, physiological anti-TDP-43 antibody and the pathology was globally mild. Our more recent cases were additionally studied with the antibody directed against phosphorylated TDP-43 protein (phospho Ser409/410, mouse monoclonal, Cosmo Bio Co, LT;1:2000).
We observed a definitely higher amount of pTDP-43 pathology than that identified using the antibody against the physiological TDP-43 protein. Similar findings have been recently reported by Josephs et al. in FTLD-TDP cases where the authors observed a higher burden of TDP-43 related neuronal lesions using this antibody compared to antibodies directed against a neoepitope in the C-terminal fragment or the N-terminal, i.e., full-length TDP-43 protein.3
In our new cases (case 1 and 2 in Table 1 with clinical details), pTDP-43 pathology was relatively extensive, and the morphology, distribution and cellular involvement were atypical for conventional FTLD-TDP subtypes. There were aberrant dense neuronal, partly filamentous and partly granular cytoplasmic inclusions in superficial and deep cortical layers, small neurites and unusual astrocytic pTDP-43 pathology (Figure 1T,Z). The astrocytic pathology varied from a fine-granular (Figure 1Z1 lower panel), p62 negative cytoplasmic immunoreactivity to p62 positive, argyrophilic fibrillar aggregates (Figure 1T, Z). Using the full-length anti-TDP-43 antibody we identified several negative astrocytic nuclei and few diffuse or dash-like inclusions, in addition to single nuclear inclusions (Figure 1Z1). Josephs et al. reported that preinclusions might be better identified with antibodies against full-length TDP-43.3 This astrocytic pTDP-43 pathology clearly followed the pathology pattern observed with the AT8 antibody at the cellular and anatomical level. Indeed, double immunofluorescence revealed co-distribution of both proteins pTau and pTDP-43, as well as of pTDP-43 and GFAP in the same cells (Figure S1A-C). Neurodegenerative changes followed an FTLD-degeneration pattern in patient 1, and involved frontal and temporal cortices, the basal ganglia, thalamus, brainstem nuclei and the cerebellar cortex. Patient 2 had clinical and neuroimaging features of predominant occipital involvement (Figure S1), but tau pathology was widespread and also affected fronto-temporal regions and was associated with hippocampal sclerosis. The type of astrocytic pTDP-43 pathology did not differ between affected anatomical areas. We also re-evaluated one of the previously reported cases by Ferrer et al. (case 3 in Table 1) where only cerebellum was available for further studies, and pTDP-43 pathology was also identified in Bergmann glia. Western blot analyses performed on frozen brain tissue of frontal cortex of the new cases 1 and 2 revealed a pTau pattern of 4R+3R isoforms. Regarding TDP-43, we observed attenuated bands in both astroglial predominant tauopathies in both, the soluble and insoluble fraction; however, increased expression levels of total TDP-43 and pTDP-43 were obtained when we used total brain homogenates (Figure S2). These findings suggest that fractionation using sarkosyl is not a useful approach to the study of TDP-43 pathology as TDP-43 species are lost particularly in cases with TDP-43 proteinopathy.
TABLE 1. Overview of clinical, neuropathological/immunohistochemical and genetic features Case number Gender Age at death Disease duration Clinical phenotype Family history 1 Female 58 6 y bvFTD Yes 2 Female 72 6 y posterior cortical atrophy No (mother died at age 53 of breast cancer and father at the age of 73 with no cognitive complaints) 3 Female 63 5 y bvFTD Yes Case number Neuropathological diagnosis Tau astroglial Tau isoforms Tau neuronal 1 FTLD-tau Widespread cortical and subcortical 4R > 3R Moderate 2 FTLD-tau with HS Widespread cortical and subcortical 4R > 3R Moderate 3 FTLD-tau with HS Widespread cortical and subcortical 4R > 3R Moderate Case number pTDP-43 glial pTDP-43 neuronalTDP-43 /
pTDP-43 CA1
TDP-43 glial TDP-43 neuronal 1 Abundant Moderate Abundant threads Isolated, negative nuclei Globally scattered, moderate in hippocampus 2 Abundant Moderate Abundant threads Isolated, negative nuclei Globally scattered 3 Abundant Moderate Yes, details n.r. Isolated, negative nuclei Globally scattered Case number FUS Trn1 TAF15 ßA4, a-synuclein 1 Negative Negative Negative Negative 2 Negative Negative Negative Negative 3 Negative n.d. n.d. Negative Case number MAPT mutation MAPT duplication Exome sequencing 1 Exon 1, 9-13 negative Absent n.d. 2 Exon 1, 9-13 negative Absent n.d. 3 Exon 2-6 and 8-12 negative n.d. FUS variant c.420G>CFurther studies are deserved to clarify the biochemical signature of TDP-43 in these cases. Recently, Neumann et al. observed differences in the immunoreaction profile between ALS-TDP / FTLD-TDP type B/C, and FTLD-TDP type A applying a newly generated antibody against TDP-43 phosphorylated at S369, suggesting the existence of distinct structural TDP-43 conformers as a substrate of the different disease phenotypes.4
No MAPT mutations or duplications were identified in cases 1 and 2. Ferrer et al. reported a FUS variation of uncertain significance in their two patients, but no abnormal aggregates of FUS have been identified in any of the cases, including those reported here. We did also not find TAF15 or Transportin1 pathology in the new cases.
There is increasing recognition of possible interactions between TDP-43 and tau proteins in neurodegeneration.5 Recently Tekeuchi et al. also reported a GGT-like pathology associated with MND and TDP-43 pathology in which Tau and TDP-43 proteins co-localized within the affected astrocytes.6 Klotz et al. also reported astrocytic TDP43 immunoreactivity accentuated in the limbic system in an elderly woman with widespread ARTAG and other concomitant proteinopathies.7 Moreover, astroglial TDP-43 has been also identified in perivascular astrocytes,8 in Cockayne Syndrome, in astrocyte-related Rosenthal fibres, and also in astrocytes of Alexander's disease,9 among other conditions,10 all showing different morphologies.
In summary, we describe prominent atypical astroglial pTDP-43 pathology in two new cases of this rare 4 > 3 repeat astroglia-predominant FTLD-tauopathy. Whether the dual pTDP-43 and pTau pathology represents a specific disease signature and whether potential protein-interactions lead to this complex pathology deserves further studies.
ACKNOWLEDGMENTSWe are indebted to the Neurological Tissue Bank of the IDIBAPS-Hospital Clinic Biobank for data and sample procurement. We are thankful to brain donors and families for generous brain donation for research. Excellent technical assistance of Sara Charif and Veronica Santiago is greatly appreciated. This study was partially supported by a grant from the “Fundació Marató de TV3” to EG (grant no. 20141610). SK holds a grant by the City of Vienna/Austria ("Hochschuljubiläumsfonds" grant number H-283459/2019).
CONFLICT OF INTERESTThe authors report no conflict of interests.
AUTHOR CONTRIBUTION1-study design, 2- acquisition of clinical data, 3-discussion and provision of neuroimaging data, 4-neuropathological work-up, 5-image acquisition, 6-genetic analysis, 7-critical discussion, 8-manuscript draft, 9-final approval of the manuscript. Gelpi Ellen: 1,4,5,7,8,9. Aldecoa Iban: 4,5,7,9. Lopez-Villegas Dolores: 2,7,9. Abellan-Vidal Maria Teresa: 2,7,9. Mercadel-Fañanas P: 2,7,9. Fortea Juan: 2,7,9. Ribosa Roser: 2,7,9. Morenas Estrella: 2,7,9. Gomez-Anson Beatriz: 3,5,7,9. Molina-Porcel Laura: 4,7,9. Ximelis Teresa: 4,7,9. Borrego Sergi: 4,7,9. Antonell Anna: 6,7,9. Rovelet-Lecrux Anne: 6,7,9. Klotz Sigrid: 4,5,7,9. Andres-Benito, Pol: 4,5,7,9. Sanchez-Valle Raquel: 7,8,9. Ferrer Isidre: 1,4,5,7,9.
ETHICAL APPROVALThe study was performed in the frame of a brain donation program in accordance with the Declaration of Helsinki. Written informed consent was obtained from patients and/or next of kin for the use of brain tissue for diagnostic and research purposes. The project was approved by the Ethics Committee of the Hospital Clinic de Barcelona, Spain.
留言 (0)