
記住我




Molecular imprinting is a means of introducing sites of specific molecular arrangement into an otherwise uniform polymeric matrix.1-3 These techniques have found applications in biomedical engineering, as chiral stationary phase in high-performance liquid chromatography, as chiral selector in capillary electrophoresis methods and in the design of new drug delivery systems.4-6 Compared to traditional molecularly imprinted polymers (MIPs) made in organic solvents, aqueous media synthesis of chemically and mechanically stable MIPs has received much traction over the past 20 years and is an interesting challenge in chemistry.7 This is due to their capability of recognising higher molecular weight molecules despite the significant reduction in integral binding strength of non-covalent template/monomer interactions. More recently, the molecular imprinting of large biomolecules, such as nucleic acids,8, 9 viruses,10, 11 and proteins,12-14 has become increasingly topical, especially with the aim of developing MIP-based sensors for the detection of disease markers. MIP-based biosensors have been reported for the determination of a number of protein biomarkers including bovine (and human) serum albumin,15-17 haemoglobin,18 myoglobin (Mb),19 and prostate-specific antigen.20, 21
The approach with biomolecular imprinting has been to use an aqueous solvent system,22 which allows the biomolecular template to remain structurally stable during and after the imprinting process. The use of water-soluble monomers and cross-linkers in the synthesis of MIPs for biomacromolecular targets is now common place. The resulting hydrogel materials are hydrophilic and highly crosslinked. Due to their high-water compatibility, hydrogel-based MIPs have been shown to retain protein stability and provide a robust means for recognition of target analytes over long periods.7, 23, 24 For hydrogel protein imprinting, water-soluble monomers such as acrylamide and functionalised acrylamides have been used alongside the cross-linker N,N′-methylenebisacrylamide (MBAm) to produce polyacrylamide-based MIPs.4, 12, 13 Polyacrylamide (PAM) is biocompatible and inert to almost any nonspecific interactions with proteins. The PAM gel monolith is processed through a 100-mesh net to produce micron-sized particles.
The constraining factor of this imprinting technology is the difficulty of the template removal. Despite that, Hawkins et al in 2005 demonstrated the efficiency of the cooperation of a strong anionic surfactant like sodium dodecyl sulphate (SDS) and acetic acid in the template removal strategy.12 Whereas this widely adopted method removes the surface exposed protein to leave protein-selective binding sites, the method is limited to exposed surfaces; any protein retained within the bulk of the microparticles remains entrapped, even after such stringent washing.
PAM materials also form the backbone of gel electrophoresis. Researchers routinely use electrophoresis to study the properties of proteins. Separation relies on charged biomolecules having different electrophoretic mobility through the PAM gel matrix because of the application of an electric field. Under constant electric field, the difference in mobility through a matrix depends on the charge and molecular weights of the molecules. Generally, the sample is run in a support matrix such as agarose or polyacrylamide gel. Agarose is mainly used to separate larger macromolecules such as nucleic acids, whereas polyacrylamide gel is widely employed to separate proteins. Slab gels, 0.5 to 1.5 mm thick, have replaced cylindrical rod gels in glass tubes because it allows direct comparison of the band pattern of different samples under identical conditions in the same matrix gel. In gel electrophoretic methods, among several other detection methods (organic dyes, fluorescent staining, and negative staining), silver staining is considered the most sensitive at low protein concentrations. All silver methods, that is, diamine or ammoniacal stains, non-diamine silver nitrate stains, silver stains based on photo-development, depend on the reduction of the ionic silver to the metallic form.
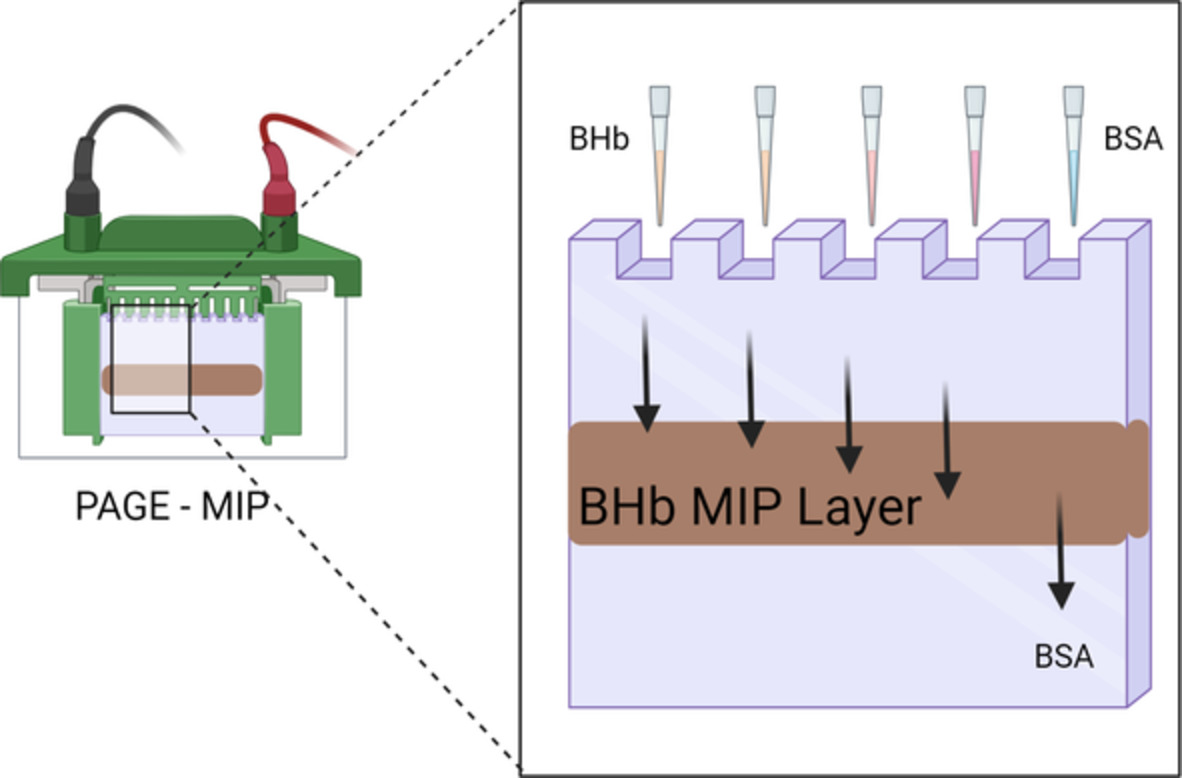
Several chromatographic applications of MIPs have been developed and applied in proteins and nucleic acids separations. Ogiso et al25, 26 applied MIP technology to gel electrophoresis to develop a simple and inexpensive DNA detection method. However, the MIP was prepared in situ in a glass tube using a specific double-standard DNA (dsDNA) target sequence as a printer molecule. What we present is the first report of hydrogel-based MIPs applied to slab electrophoresis, involving a multilayer resolving system. Illustrated is the application of hydrogel-based MIPs in mini-slab gel electrophoresis whereby a MIP dispersion is layered between two control layers. Using the three-layered mini-slab, a non-imprinted polymer (NIP) dispersion instead of MIP as a control system was also investigated. In this article, we explore how, during electrophoresis, a protein imprinted polymer can retain its template protein vs a protein, which is analogous in molecular weight.
2 MATERIALS AND METHODS 2.1 MaterialsAcrylamide (AA), N,N-methylenebisacrylamide (MBAm), ammonium persulphate (APS), N,N,N,N-tetramethylethyldiamine (TEMED), sodium dodecyl sulphate (SDS), glacial acetic acid (AcOH), Tris(hydroxymethyl)aminomethane (Trizma base), bovine haemoglobin (H2500, BHb), and bovine serum albumin (A3059, BSA) were all purchased from Sigma-Aldrich (Poole, UK). Glycine, sodium hydroxide, silver nitrate, methanol, sodium thiosulfate, 2-propanol bromophenol blue, glycerol, sodium carbonate anhydrous, and formaldehyde solution 37% to 41% were all purchased from Fisher Scientific (Loughborough, UK). Sieves (75 μm) were purchased from Inoxia Ltd. (Guildford, UK).
2.2 Electrophoresis apparatusA BioRad Mini Protean 3 gel apparatus was used in the preparation of polyacrylamide gel slabs and the electrophoresis experiments. The dimensions of the mini-slab gels for this BioRad apparatus were 7.3 × 8 cm2. The two spacers were positioned by placing the Teflon sheet (supplied with the BioRad Mini gel apparatus) between the spacers. Combs with the same thickness containing 10 slots were used to form gel wells. Compared to standard gel systems, the mini system minimises reagent consumption and reduces electrophoretic run time. Assembly of the glass plates to form the gel mould was achieved using two one-piece clamps. The clamps held the glass plates apart by the required distance, and the sandwich was then locked onto the casting stand. The gelling solution was then poured. After polymerisation, the gel sandwich was transferred from the casting stand to the upper buffer chamber. The entire inner glass plate of the gel sandwich was in contact with the upper buffer, creating even heat distribution for “smile-free” separations.
2.3 Solution preparationsA previously established template elution method employing 10% (vol/vol) solution of AcOH containing 10% (wt/vol) SDS (pH 2.8) was used to remove template from the MIP.12 Glycine 0.4 M stock solution was employed as both resolving gel buffer and to prepare the sample solutions. These were adjusted to pH 8.8 using sodium hydroxide 1 M and then diluted to by 1:2. A Trizma base (0.06 M) and glycine (0.48 M) solution were used as running buffer solution (pH 8.4). AA/MBAm 30% total density (T, wt/vol) and 2.6% crosslinking density (C, 37.5:1, wt/wt) were filtered through a 0.38-μm membrane filter. The sample buffer was prepared using 25% (vol/vol) resolving buffer, 20% (vol/vol) glycerol, and 5% (vol/vol) of bromophenol blue solution prepared from 0.1% (wt/vol) stock solution. In the silver-staining procedure, a solution of H2O: methanol: acetic acid (50:40:10) was used as fixer solution. For sensitising the gel, a 0.03% (wt/vol) sodium thiosulfate solution was used. A 0.1% (wt/vol) silver nitrate solution at 4°C was employed in the silver-staining procedure. For the reduction of silver ions to metallic silver a 0.04% (vol/vol) formaldehyde in 2% (wt/vol) sodium carbonate solution was used. To halt the staining process, the gels were soaked in an acetic acid solution 5% (vol/vol).
2.4 MIP fabrication and conditioningTemplate BHb (12 mg, 186 μM), AA (54 mg, 0.76 M), MBAm (6 mg, 38.9 mM), 20 μL of 10% (wt/vol) APS (as initiator, 8.77 mM), and 20 μL of 5% (vol/vol) TEMED (as catalyst, 8.61 mM) were mixed in reverse osmosis (RO) water to give a final volume of 1 mL. After nitrogen degassing for 5 minutes, free radical polymerisation of the gel occurred overnight at room temperature (~22°C) giving final gel densities of 6% T (wt/vol) and 10% C (9:1, wt/wt). Molar ratios of monomer and cross-linker to template protein were at 4086:1 and 209:1 respectively. A non-imprinted polymer (NIP) control system was prepared using the same method but in the absence of template BHb.
After polymerisation, the gels were granulated separately using a 75-μm sieve. Of the resulting gels, 0.1 g was transferred to 1.5 mL polypropylene Eppendorf tubes and washed with five 0.2 mL volumes of RO water followed by five 0.2 mL volumes of SDS:AcOH eluent12 and five 0.2 mL volumes of RO water again to remove any residual SDS:AcOH eluent and equilibrated the gels. Each wash/elution/wash step was followed by centrifugation using an Eppendorf mini-spin plus centrifuge for 3 minutes at 6000 rpm (RCF: 2419g). All supernatants were collected for spectrophotometric analysis using a UV mini-1240 CE spectrophotometer at 404 nm for BHb (Shimadzu Europa, Milton Keynes, UK) to verify the extent of template removal. It should be noted that the last water wash and SDS:AcOH eluent fractions were not observed to contain any protein. Therefore, we are confident that any remaining template protein within the MIPs did not continue to leach out during subsequent studies.
2.5 MIP characterisation and selectivityThe subsequent rebinding effect and selectivity of the conditioned and equilibrated BHb-MIPs and NIPs were characterised using spectrophotometry for their affinity towards template BHb and cognate BSA using single-point analysis. Hydrogel MIPs and NIPs (0.1 g) were treated with a 3 mg/mL protein solution (either template BHb or cognate BSA) prepared in 0.2 mL RO water, and polymer/protein solutions were mixed on a rotary vortex mixer then allowed to associate at room temperature (~22°C) for 20 minutes followed by centrifugation. The hydrogels were then washed four times with 0.2 mL RO water. Each reload and wash step for the hydrogels was followed by centrifugation, and all supernatants were collected for analysis by spectrophotometry at 404 nm for BHb and 280 nm for BSA, using a UV mini-1240 CE spectrophotometer (Shimadzu Europa, Milton Keynes, UK).
2.6 Multilayer mini-slab hydrogel productionFirstly, a 1.2 mL solution consisting of 960 μL of resolving buffer, 500 μL of AA/MBAm mixtures solution (30% T, 2.6% C), 20 μL of APS 10% (wt/vol) solution, and 20 μL of TEMED 5% (vol/vol) was poured into the space between the two glass plates of the polyacrylamide gel electrophoresis (PAGE) apparatus. Hence, the final concentration of the gel was 10% (wt/vol) and represented the bottom layer of the gel. As soon as the solution was poured, the gel was layered with a few drops of 2-propanol to flatten the layer. After 10 minutes, the 2-propanol drops were drained away and the second layer was applied. This comprised 0.1 g of the preconditioned and equilibrated MIP (or NIP for the control system) dispersed in 1.5 mL of an identical solution to that of the first (bottom) layer. Thus, 1.2 mL of that dispersion was poured as a second layer into the space in between the two plates, and the same procedure as the first layer was followed. The third and final layer was filled using an identical solution as the first layer. The three-layered gel polymerising was then left for at least 4 to 5 hours before injecting the samples.
2.7 Native polyacrylamide gel electrophoresisBHb or BSA protein samples (500-4000 ng) were prepared in sample buffer solution to give a final volume of 50 μL, and then, the solutions were loaded directly onto the sample wells. After loading the samples, the gel was run at 150 V at different run times. The gels were then carefully removed using a blade and the European Molecular Biology Laboratory (EMBL) silver-staining protocol was followed in staining the gels.27
3 RESULTS AND DISCUSSION 3.1 MIP characterisation Table 1 illustrates the molecular imprinting effects of a BHb-MIP in recognising its original template BHb and non-cognate BSA in relation to a NIP control. These have been characterised by calculating the rebinding capacity (Q, mg/g) of proteins to the gel polymer using: (1)where Ci and Cr are the initial protein and the recovered protein concentrations (mg/mL), respectively (which specifies the specific protein bound within the gel), V is the volume of the initial solution (mL), and g is the mass of the gel polymers (g).The imprinting factor (IF) servers as a standard and is expressed by comparing the latter calculated binding capacities (Q) for MIP and NIP control (Equation 2):
(1)where Ci and Cr are the initial protein and the recovered protein concentrations (mg/mL), respectively (which specifies the specific protein bound within the gel), V is the volume of the initial solution (mL), and g is the mass of the gel polymers (g).The imprinting factor (IF) servers as a standard and is expressed by comparing the latter calculated binding capacities (Q) for MIP and NIP control (Equation 2):
 (2)
TABLE 1.
Characterisation of the imprinting effect of bovine haemoglobin (BHb) using rebinding capacities (Q), imprinting factors (IF), and relative imprinting factors (k) for a BHb-imprinted MIP towards its native BHb template and cognate BSA
Q (mg/g)
IF
k
% Rebinding efficiency
Analyte
BHb-MIP
4.79 ± 0.12
8.1
1
70%
BHb
NIP
0.59 ± 0.09
BHb-MIP
3.95 ± 0.17
4.2
0.52
50%
BSA
NIP
0.95 ± 0.32
Note: Data collected using single-point analysis, representing mean ± SEM, n = 3.
The selectivity of the BHb-imprinted MIPs for cognate proteins was quantified using relative imprinting factors (k; Equation 3):
(2)
TABLE 1.
Characterisation of the imprinting effect of bovine haemoglobin (BHb) using rebinding capacities (Q), imprinting factors (IF), and relative imprinting factors (k) for a BHb-imprinted MIP towards its native BHb template and cognate BSA
Q (mg/g)
IF
k
% Rebinding efficiency
Analyte
BHb-MIP
4.79 ± 0.12
8.1
1
70%
BHb
NIP
0.59 ± 0.09
BHb-MIP
3.95 ± 0.17
4.2
0.52
50%
BSA
NIP
0.95 ± 0.32
Note: Data collected using single-point analysis, representing mean ± SEM, n = 3.
The selectivity of the BHb-imprinted MIPs for cognate proteins was quantified using relative imprinting factors (k; Equation 3):
 (3)where IFtemplate is the imprinting factor for the original template, and IFanalogue is the imprinting factor of the analogue proteins. For the template BHb, k = 1, and for the non-cognate proteins that are less specific for the BHb-MIP, k Q = 4.79 and 3.95 mg/g, respectively). BSA has also been expressed as having a 0.52 k to a BHb-MIP, meaning that more BHb is specifically bound by our MIP. Thus, our BHb-MIP has more recognition for BHb, in terms of selectivity and affinity, than non-cognate BSA when analysed spectrophotometrically for bulk gel imprinting prior to PAGE application.
3.2 Native protein polyacrylamide gel electrophoresis
(3)where IFtemplate is the imprinting factor for the original template, and IFanalogue is the imprinting factor of the analogue proteins. For the template BHb, k = 1, and for the non-cognate proteins that are less specific for the BHb-MIP, k Q = 4.79 and 3.95 mg/g, respectively). BSA has also been expressed as having a 0.52 k to a BHb-MIP, meaning that more BHb is specifically bound by our MIP. Thus, our BHb-MIP has more recognition for BHb, in terms of selectivity and affinity, than non-cognate BSA when analysed spectrophotometrically for bulk gel imprinting prior to PAGE application.
3.2 Native protein polyacrylamide gel electrophoresis
In order to study how selectively and specifically a MIP binds a certain molecule during electrophoretic procedures, we set the experiments using a multilayer system in mini-slab gel electrophoresis where a dispersed MIP layer with binding sites available was in between other two “non-imprinted” gel layers. We used an imprinted polymer, based on polyacrylamide hydrogels for the selective imprinting of bovine haemoglobin (BHb, MW 65 kDa) as a discriminating layer able to specifically bind BHb molecules instead of other proteins similar in molecular weight, namely, albumin from bovine serum (BSA, MW 66 kDa). A three-layer mini-slab with a NIP dispersion instead of a MIP in the middle layer was considered as a control system. Resulting stains are shown in Figures 1-3. In the first set of experiments, we explored the critical amount of haemoglobin that was detectable in the multilayer system configuration. Figure 1 illustrates that the layer containing the MIP exhibits significant staining throughout the gel. This is due to the fact that the MIP particles used have residual haemoglobin locked within the gel particles, which has therefore also been stained. Tracks a-c in the electrophoresis experiment in Figure 1 show no breakthrough of protein into the bottom layer, which confirms that the firstly, the inaccessible protein within MIP particles does not leach out and that injected concentrations of 500 to 2000 ng are not transported to the bottom layer during electrophoresis. This suggests that the MIP-loaded middle layer is retaining up to 2000 ng of protein even under an electrophoretic field for 120 minutes. In contrast, when the NIP is in the middle layer (Figure 1), we observe breakthrough of protein into the bottom layer at 1000, 2000, and 4000 ng (tracks f, g, and h, respectively), but little to no breakthrough at 500 ng (track e). This demonstrated that while the NIP was not able to retain protein, the MIP could selectively bind 500 to 2000 ng of protein.

PAGE study using increasing quantities of bovine haemoglobin (BHb) in BHb-MIP (a-d) and non-imprinted polymer (NIP) control systems (e-h): 500 ng in tracks a and e, 1000 ng in tracks b and f, 2000 ng in tracks c and g, 4000 ng in tracks d and h. Run time = 120 minutes. Arrow indicates the direction of protein migration

Run time effects using 4000 ng BHb in BHb-MIP (tracks a-c) vs NIP (tracks d, e). In tracks a and d, the run time is 120 minutes, while in b, c, and e, the run time is 150 minutes. Arrow indicates the direction of protein migration

Selectivity study using a 120 minutes run time and 4000 ng of protein: BSA in BHb-MIP (a) and NIP control (c); BHb in BHb-MIP (b); and NIP control (d). Arrow indicates the direction of protein migration
The three-layer system for MIP- and NIP-loaded middle layers was investigated further at 4000 ng BHb injection, and a comparison was made between electrophoresing at 120 and at 150 minutes (see Figure 2). These high BHb loadings confirmed that both the MIP and NIP layers can retain BHb in the initial 120-minute period. Tracks a and d (Figure 2) represent MIP and NIP, respectively, at an electrophoretic run time of 120 minutes, and tracks b, c (MIP), and e (NIP) represent BHb retention or release requiring a further 30 minutes of electrophoresis (total 150 minutes). In track e (NIP), the exited protein band is observed throughout the bottom layer. Conversely, in tracks b and c (MIP), almost all of the protein remains trapped within the MIP (middle) layer. This is evident by the lack of stained protein within the bottom gel layer of those tracks.
Furthermore, to explore the selectivity of the imprinted cavities, 4000 ng of either BHb or BSA was tested against the three-layer MIP-PAGE system. Figure 3 illustrates MIP and NIP dispersion layers showing similar behaviours towards BSA (tracks a and c). BSA in both gels appears in three different bands with dissimilar electrophoretic mobility. A possible explanation could involve heterogeneity of the gel's interaction with BSA molecules forming water-soluble covalent conjugates,28 or perhaps denaturing of the protein in the set conditions with the synthesis of dimers or aggregates.29, 30 Despite this, the BSA pattern in gel electrophoresis is clear in that BSA bands exhibit an identical distance of migration in both MIP and NIP systems. The most significant evidence of non-interaction between BSA molecules and MIP cavities can be noticed considering the BSA band is clearly present in the bottom layer of both the MIP and NIP gel systems. In both the MIP and NIP systems, the BSA band covers the same distance, while the BHb bands (tracks b and d in Figure 3) are almost totally (selectively) retained within the discriminating MIP. The latter results therefore demonstrate selectivity of MIPs for the template BHb molecule and not BSA. Whereas the two native proteins are of similar molecular weight and pI, they differ in molecular shape and conformation.
All experiments in our study were conducted at 150 V, which is typically the upper voltage limit used for conventional protein gel electrophoresis (typically 100-150 V). It is likely that higher applied voltages would impact the recognition ability of the MIP, but we did not see it necessary to study voltages higher than 150 V. The recognition ability was defined in this study as the ability of the MIP layer to retain the target protein in contrast to non-target protein (demonstrated in Figure 3) or the corresponding control (NIP) polymer, which is the case when comparing Figure 1 (track d) against Figure 1 (track h). Effectively, the MIP selectively halts the migration of the target protein. Eventually, a longer run time (beyond 150 min) may take precedence and the protein could be electro-eluted from the MIP layer despite the MIP's recognition ability.
These results will inform further studies concerning the MIP-based separation and identification of proteins important in diagnosing diseases. Our approach paves the way for the development of hydrogel-based MIPs, which can selectively trap and separate one protein over others in a process that requires less than 2.5 hours. In this multilayer configuration, it could be possible to inject a sample of different proteins where the MIP system will selectively trap only one well-defined molecule. We can then tailor the run time to release and discard nonspecific proteins.
4 CONCLUSIONSThe binding of a BHb-imprinted MIP to its native template and non-cognate BSA protein has been assessed using gel electrophoresis. Both spectrophotometry and PAGE methods demonstrate imprinting and selectivity towards template BHb over cognate BSA. The results show how an imprinted polymer retains its specific protein molecule within its cavities even under the influence of an electric field during electrophoresis. The application of MIPs within slab electrophoresis could further aid in the separation of similar sized proteins.
ACKNOWLEDGEMENTThe authors wish to thank the British council—Erasmus programme and the Natural Environment Research Council with the Royal Society of Chemistry (NE/J01/7671) for supporting this work. The authors also gratefully acknowledge the assistance of Jane A. Newcombe, University of Surrey and Luigi Campanella, University of Rome.
CONFLICT OF INTERESTThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONSFlavio Giosia, Hazim F. EL Sharif, and Subrayal M. Reddy contributed to the conception and design of the study. Flavio Giosia and Hazim F. EL Sharif performed the study and analysis. Subrayal M. Reddy, Flavio Giosia, and Hazim F. EL Sharif wrote the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
留言 (0)