
記住我


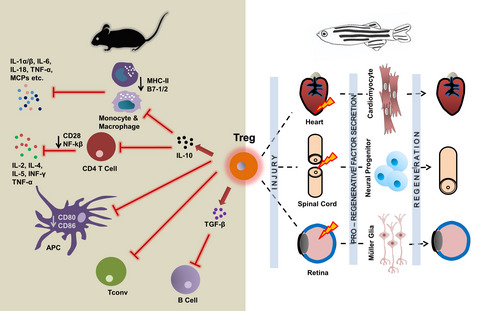




Microglia are the resident immune cells of the central nervous system (CNS). They originate from the yolk sac where progenitors, during development, migrate and populate the brain [1, 2]. Following this migration, the microglia population is maintained during the life-span by self-renewal through apoptosis and proliferative processes [3]. During development, microglia play a key function in shaping the neuronal network by supporting neural development and synapsis pruning [4, 5], while in the adult CNS they carry surveillance functions to maintain homeostasis. Taking advantage of their ramified plastic morphology and their high mobility, they continuously sense their environment for pathological insults and harmful stimuli [6]. Microglia are the first line of defense in the brain. In response to injury and disease pathogens they undergo cellular activation characterized by morphological change (from ramified into amoeboid shape), gliosis and functional changes, including alteration of cell surface receptors expression and production of cytokines and chemokines. Depending on the nature of the stimuli, activated microglia have been largely classified in two phenotypes, namely pro-inflammatory and anti-inflammatory, sitting at opposite ends of the activation spectrum. Pro-inflammatory microglia are generally described as producers of inflammatory mediators, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, macrophage inflammatory protein (MIP)-1 α, nitric oxide synthase (NOS) and reactive oxygen species (ROS), via which they exert their detrimental effects [7]. Conversely, anti-inflammatory microglia are involved in dampening the inflammatory response and promoting the repair mechanisms. They are characterized by secretion of anti-inflammatory cytokines such as IL-4, IL-10, IL-13 and transforming growth factor (TGF)-β and up-regulation of markers such as arginase-1 (Arg-1), FIZZ-1, CD206 and YM-1 [7]. However, recent single-cell transcriptomics and proteomics studies disputed this pro-/anti-inflammatory dichotomy and identified a multitude of activated microglia phenotypes due to a variety of gene signatures associated with diseases and depending on their environmental context [8-10].
Despite this tremendous progress in identifying the different microglia phenotypes in vitro and in vivo, a clear understanding of these processes in the human brain in vivo is still lacking. A non-invasive imaging technique such as positron emission tomography (PET) is well suited for quantitative investigation of neuroinflammation and possesses the potential to discriminate between the different microglia phenotypes. Although it is now appreciated that the simple pro- and anti-inflammatory phenotype may not capture the whole diversity of microglia activation [11], this classification serves as a crucial guide for PET tracer development. In this context, we understand the pro-inflammatory response to be associated with disease progression and neurotoxicity, while the anti-inflammatory response is linked to dampening of harmful effects and recovery. Most of the development in PET imaging of neuroinflammation has focused upon targeting translocator protein 18 kDa (TSPO). Despite the large panel of tracers developed, TSPO appeared to be a less than ideal target due to, among others, its expression on different brain cells and the lack of ability to differentiate between pro- and anti-inflammatory microglia. These drawbacks encouraged the investigation of other targets that might be more selective for microglia and can differentiate between the activated phenotypes.
In the current review, we first summarize the different microglia activation phenotypes in the main chronic neuroinflammatory and neurodegenerative diseases, focusing on multiple sclerosis (MS), Alzheimer’s disease (AD) and Parkinson’s disease (PD). Then, we discuss the current and emerging PET imaging targets and their tracers and highlight, when characterized, their potential in discriminating between microglia activation phenotypes.
Microglia activation phenotypes in neuroinflammatory and neurodegenerative diseasesNeuroinflammation is an inflammatory process within the CNS and occurs as response to various pathological insults, such as infection, protein aggregation, trauma and ischemia. It is mainly driven by microglia and astrocytes and in certain cases by infiltrated immune cells from the blood, especially when the blood–brain barrier (BBB) is inflamed and compromised. The secretion of pro-inflammatory cytokines and chemokines, nitric oxide (NO) and ROS usually mark this process. In an acute event, the mounted neuroinflammatory response will resolve when the triggering insults are eliminated, and microglia will transition into an anti-inflammatory and regenerative phenotype. However, in neurodegenerative diseases, neuroinflammation tends to become a chronic process that fails to resolve itself due to continuous stimulation. This chronic activation leads to a variety of microglia phenotypes that potentially contributes to neuronal damage and death and becomes a major driver of disease progression.
In this section we concisely highlight the activation phenotypes of microglia in neuroinflammatory and neurodegenerative diseases and how they contribute to the disease pathology, focusing upon MS, AD and PD (Table 1).
TABLE 1. Activated microglia phenotypes and the expressed cytokines and markers in neuroinflammatory and neurodegenerative diseases Disease Microglia phenotype Expression of markers and cytokines Reference Multiple sclerosis Initial and early white matter active lesions Pro-inflammatory CD68, MHC-I, MHC-II, CD86 [14] ROS, p22phox Late active white matter lesions Intermediate phenotype MIP-1β (CCL4), OPN [15-17] HLA-DR, CD11c, AXL, CD45, CD68, CD206, CD163 Slowly expanding white matter lesions (active rim) Pro-inflammatory CD40, CD68, p22phox, iNOS, ferritin [14, 18] IL-1, IL-6 Cortical lesions MS 1 ↓ P2Y12R, ↑HLA-II, ↑CD68 [21] MS 2 ↓ P2Y12R, hyper-ramified morphology [21] EAE animal model (early phase) Pro-inflammatory CD86, CD40, MHC-II [22] TNF-α, IFN-γ, IL-12, IL-6, iNOS, NO EAE animal model (recovery phase) Anti-inflammatory IL-4, IL-10 and IL-13 [22, 23] Alzheimer’s disease AD (late stage) Pro-inflammatory IL-1β, TNF-α, p40, MHC-II, iNOS [9, 31] AD1 (associated with Aβ) ↓ P2Y12R [10] ↑ TREM2, ↑ ApoE4, ↑ITGAX AD2 (associated with pTau) ↑ GRID2, P2Y12R, ↑CX3CR1 [10] AD transgenic mice Anti-inflammatory (early Aβ pathology) YM1 [30] Dynamic shift towards pro-inflammatory (later stage) DAM (phagocytic) ↓ P2y12, ↓ CX3CR1, ↓ TMEM119 [32] ↑ ApoE4, ↑ Tyrobp, ↑TREM2 Dark microglia CD11b, TREM2 [39] Parkinson’s disease PD Pro-inflammatory COX-2, iNOS [43, 44] IL-1β, IL-6, TNF-α PD animal models Pro-inflammatory IL-1β, IL-6, TNF-α, NOS2 [55] ↓ IGF-1, ↓ MRC1 Abbreviations: ↑, Over-expression; ↓, down-regulation; AD, Alzheimer’s disease; ApoE4, apolipoprotein E-4; CX3CR1, CX3C chemokine receptor-1; EAE, experimental autoimmune encephalomyelitis; GRID2, glutamate receptor, ionotropic, delta 2; HLA-DR, human leukocyte antigen D-related; IFN, interferon; IGF, insulin-like growth factor; IL, interleukin; iNOS, inducible nitric oxide synthase; ITGAX, integrin subunit alpha X; MHC, major histocompatibility complex; MIP, macrophage inflammatory protein; MRC, mannose receptor C; NO, nitric oxide; OPN, osteopontin; P2Y12R, purinergic 2Y receptor type 12; ROS, reactive oxygen species; TNF, tumor necrosis factor; TREM, triggering receptor expressed on myeloid cells; Tyrobp, transmembrane immune signaling adaptor. Multiple sclerosis (MS)MS is a chronic inflammatory disease of the CNS. The main pathological hallmark of MS is the presence of focal lesions in the white matter associated with extensive glia activation, demyelination and neurodegeneration [12, 13]. Activated microglia are at the core of the neuroinflammatory response in MS pathology. Recent discoveries of specific markers and molecular signatures of microglia [transmembrane protein 119 (TMEM119) and purinergic 2Y receptor type 12 (P2Y12R)] indicated their predominance over monocyte-derived macrophages in MS lesions [14, 15]. Active demyelination and axonal injury are associated with the focal accumulation of activated microglia; however, the dynamics of microglia activation and their phenotypes in association with lesion types and disease progression is still not well understood. In white matter initial and early active lesion stages, microglia predominantly showed a pro-inflammatory phenotype with expression of phagocytic and antigen presentation markers [CD68, major histocompatibility complex (MHC) classes I and II and CD86] and production of ROS and enzymes that produce them (p22phox). In later stages of active lesions and inactive microglia-containing cores of active lesions, microglia adopted an intermediate phenotype and co-expressed anti-inflammatory markers (CD206 and CD163) together with pro-inflammatory markers [14, 16, 17]. Single-cell mass cytometry analysis on active lesions of progressive MS revealed enriched clusters of cells with higher expression of phagocytosis-related markers [human leucocyte antigen D-related (HLA-DR), CD11c, AXL, CD45, CD68] and inflammatory molecules such as inflammatory cytokines MIP-1β (CCL4) and osteopontin (OPN) [15]. In slowly expanding lesions, microglia present at the active rim adopted mainly a pro-inflammatory phenotype with expression of CD40, CD68, p22phox, inducible nitric oxide synthase (iNOS) markers and ferritin, while expression of anti-inflammatory markers was very low [14, 18]. This phenotype was supported by data from RNA microarrays on micro-dissected tissues showing strong expression of complement factors and interleukins (IL-1 and IL-6), which play a role in pro-inflammatory processes [18]. It is evident that pro-inflammatory microglia contribute to the demyelination and axonal injury in the early active lesions and the active rim of chronic lesions [19]. The increased expression of antigen-presenting markers, inflammatory cytokines and ROS support their strong role in the development of MS pathology. Active remyelination in MS lesions is most prominent in active white matter lesions, which harbor a high content of microglia with intermediate phenotype. In contrast, no or minimal remyelination is seen in slowly expanding lesions [20] where mainly pro-inflammatory microglia are present. Cortical lesions are also present in MS; a recent study identified the presence of two distinct MS-specific microglia in these lesions named MS1, defined by a low expression of P2Y12R, high HLA-II and CD68 and MS2 characterized by a hyper-ramified morphology with low P2Y12R expression [21]. Interestingly, only MS2 microglia were associated with neuronal loss in the cortical lesions [21].
Experimental autoimmune encephalomyelitis (EAE) is the most used model to mimic the pathology observed in MS. Pro-inflammatory microglia are predominantly present in the early phase of EAE and express high levels of CD86, CD40 and MHC-II on their surface. The prominent expression of pro-inflammatory cytokines such as TNF-α, interferon (IFN)-γ, IL-12, IL-6, iNOS, NO and proteases highlight their important role in the early phase of EAE development [22]. The level of anti-inflammatory microglia increases with EAE progression and peaks at the maximum of the clinical symptoms and in the recovery phase at the expense of pro-inflammatory microglia. This dynamic shift towards anti-inflammatory microglia dominance and the release of a variety of cytokines such as IL-4, IL-10 and IL-13 play an important role in the resolution of inflammation and repair mechanisms [22, 23]. This pro- and anti-inflammatory polarization balance highly correlates with the demyelination/remyelination process in acute demyelination models, probably via modulation of the inflammatory niche and oligodendrocyte progenitor cell response [24, 25].
Microglia activation in MS shows a very complex spatial and temporal pattern, and the exact role and contribution of microglia phenotypes to the evolution of MS lesions at different stages of the disease is still not totally clear.
Alzheimer’s disease (AD)AD is the most common type of neurodegenerative diseases of the CNS and is associated with progressive cognitive decline and memory loss. Pathologically, AD is characterized by extracellular deposition of amyloid beta (Aβ) plaques and intraneuronal accumulation of phosphorylated tau tangles [26]. Neuroinflammation is a prominent feature of AD. Microglia have been identified around Aβ plaques and shown to undergo dramatic morphological and electrophysiological changes in comparison to plaque-distant microglia, indicating an activated state [27, 28]. In AD, microglia are able to bind soluble Aβ and Aβ fibrils via cell-surface and Toll-like receptors, and microglia phagocytic activity of Aβ has been identified in vitro and in vivo, exhibiting their role in Aβ clearing [29]. Microglia surrounding the plaques express YM-1 and are thus suggested to adopt an anti-inflammatory activation phenotype [30]. An age-related dynamic shift of microglia phenotype is reported in the PS1M146LxAPP751SL double mutated mice. Anti-inflammatory microglia with phagocytic characteristics at the beginning of Aβ pathology could switch to a pro-inflammatory phenotype at the advanced stage of disease in this model, which is induced in part by the age-dependent accumulation of soluble Aβ oligomers [30]. Activated microglia in a later AD stage showed an increased expression of pro-inflammatory markers such as IL-1β, TNF-α, p40, MHC-II and iNOS, but not the anti-inflammatory factors IL-4 or IL-10 [9, 31].
Recent transcriptomics studies identified a microglia phenotype associated with neurodegenerative diseases, termed disease-associated microglia (DAM) [32]. DAM stained positively for intracellular Aβ particles and are spatially localized in the proximity of Aβ plaques in the 5XFAD mice, a transgenic animal model of AD, and in human post-mortem brain [32]. DAM demonstrated significant changes in gene expression, including down-regulation of homeostatic genes such as P2y12, CX3C chemokine receptor-1 (Cx3cr1) and TMEM119, and up-regulation of known AD risk factors such as apolipoprotein E-4 (ApoE4), transmembrane immune signaling adaptor (Tyrobp) and TREM2 [32, 33]. Triggering receptor expressed on myeloid cells 2 (TREM2) is a microglia-specific cell surface receptor, and loss-of-function mutations in TREM2 are associated with increased risk of developing AD [34]. TREM2 binds Aβ and works as a sensor molecule for microglia activation [35]. TREM2-deficient microglia showed reduced uptake of Aβ-lipoprotein complexes in vitro and minimal evidence of Aβ internalization in vivo [36, 37]. The stimulation of TREM2 initiates signal transduction pathways that promote microglial chemotaxis, phagocytosis, survival and proliferation, suggesting an anti-inflammatory and protective role of DAM in AD [34, 38]. A new phenotype of microglia, referred to as ‘dark microglia’, was identified in the APP/PS1 AD mouse and demonstrated a highly activated state with strong expression of CD11b and TREM2 [39]. Microglia activation in AD is complex, and results in a large variety of phenotypes [9] due to the presence of different stimuli and variety of activation pathways. Microglia activation and phenotypes evolve as a function of disease progression. A loss of homeostatic signature and transition of microglia into DAM population is a function of disease progression and occurs in two phases with a TREM2-independent initial step and TREM2-dependent second step [9, 32]. Recently, in a snRNAseq study on human AD tissues, two distinct AD-associated microglia profiles were identified that associated either with Aβ (termed ‘AD1’) or with hyper-phosphorylated tau (termed ‘AD2’) [10]. AD1 microglia correlated with phagocytic/activated markers including ITGAX, LPL, GPNMB, MYO1E and SPP1, while AD2 microglia were enriched with homeostasis genes such as CXCR1, P2Y12R and neuron-related genes such as GRID2, ADGRB3 and DPP10 [10]. Interestingly, AD1 microglia showed a similar gene expression profile as the DAM identified in the amyloid mice model [32] with increased expression of TREM2 and APOE and down-regulation of P2Y12R [10].
Current evidence suggests that at the early stages of AD, microglia have a protective role marked by phagocytosis and clearance of Aβ and secretion of anti-inflammatory cytokines. However, as AD progresses, chronic stimulation and activation cause phenotypical changes in microglia towards a pro-inflammatory phenotype. The pro-inflammatory environment can alter the phagocytic activity of microglia, which possibly leads to increased accumulation of Aβ [38], and can stimulate neurotoxic astrocytes via IL-1α and TNF [40]. In addition, complement-mediated (C3R) synapse phagocytosis and loss induced by pro-inflammatory microglia contribute to neurodegeneration and disease progression [38].
Parkinson’s disease (PD)Progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of intraneuronal Lewy body inclusions and neurites containing protein aggregates are the main pathological hallmarks of PD [41]. In PD patients, dopaminergic neuronal death results in progressive motor symptoms, including tremor at rest, bradykinesia, rigidity and postural instability. During the last decade it has become evident that neuroinflammation is associated with PD and activated microglia are present in the SNpc of PD patients [42]. In post-mortem PD tissues, activated microglia express high levels of inflammatory cytokines such as IL-1β, IL-6 and TNF-α, as well as iNOS and cyclooxygenase-2 (COX-2), suggesting a pro-inflammatory activated phenotype [43, 44]. Analysis of cerebrospinal fluid (CSF) and peripheral blood plasma from PD patients also showed elevated levels of IL-1β, IL-6 and TNF-α [44-46]. These inflammatory cytokines were also increased in the 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) models of PD, while anti-inflammatory cytokines and markers such as TGF-β and CD206 were decreased [47, 48]. In non-human primates, long-term glial activation and high levels of TNF-α and IFN-γ were detected several years following MPTP treatment [49]. Importantly, the amounts of IFN-γ and TNF-α in the SNpc correlated with the degree of neurodegeneration and motor impairment [49]. The exposure of dopaminergic neurons to inflammatory cytokines has been shown to promote neuronal death. Over-expression of TNF-α or IL-1β, pro-inflammatory microglia cytokines, in the SNpc using an adenovirus vector was sufficient to induce nigral neuronal degeneration [50, 51]. However, neutralization of soluble TNF by intranigral administration of engineered dominant negative TNF compound XENP345 reduced the retrograde nigral degeneration in the 6-OHDA model by 50% [52]. This emphasizes the cardinal role of pro-inflammatory microglia and cytokines in dopaminergic neuronal degeneration. Another feature of PD pathology is the presence of α-synuclein (α-syn) aggregates that constitute the main component of the Lewy bodies and neurites. Extracellular α-syn oligomers function as damage-associated molecular patterns. Signaling via Toll-like receptor (TLR)-2 and -4, α-syn activates microglia into a pro-inflammatory phenotype and induces the secretion of inflammatory cytokines and markers such as TNF, IL-1β, IL-6, COX-2, NOS and ROS [53, 54]. Administration of preformed fibrils in mouse brains induced the expression of pro-inflammatory markers by microglia including NOS2, IL-6, TNF and IL-1β without affecting or decreasing anti-inflammatory markers such as IFN regulatory factor-4 (IRF-4), insulin-like growth factor-1 (IGF-1) and mannose receptor C-1 (MRC-1) [55]. Additionally, activated microglia play an active role in the process of α-syn transmission to neurons via exosomes that is further amplified by the presence of pro-inflammatory cytokines [56].
The role of pro-inflammatory microglia as initiator or secondary effector in PD is still not clear. Chronically activated microglia and the secretion of high levels of pro-inflammatory mediators that damage the neurons and induce the release of α-syn further activate microglia resulting in a feed-forward effect, promoting inflammation and neurodegeneration.
Translocator protein 18 kDa (TSPO) – gold standard for PET imaging of activated microgliaPET imaging research of microglial activation and neuroinflammation has been traditionally focused upon TSPO. TSPO is predominantly expressed on the outer mitochondrial membrane in multiple cell types in periphery and CNS and is linked to several critical cellular functions, including inflammatory pathways. TSPO PET, particularly using (R)-[11C]PK11195, is often referred to as the gold standard for PET imaging of neuroinflammation. Limitations of (R)-[11C]PK11195, including the low signal-to-noise ratio, have led to an extensive search for a more optimal TSPO radioligand, both taking into account an increase in affinity to reduce non-specific binding (second-generation TSPO ligands) and the difference in ligand binding affinity induced by single-nucleotide polymorphism rs6971 (third-generation TSPO ligands), reviewed in detail recently [57, 58]. Next to limitations of the PET ligands, targeting TSPO for neuroinflammation has challenges and limitations in itself. First, knowledge on levels and cellular localization of TSPO expression in healthy and diseased human brain largely stems from post-mortem studies using TSPO radioligands, but results are highly inconsistent [59, 60]. Cellular sources of TSPO expression in healthy and diseased brain have recently been reviewed by Nutma et al. [61]. In short, immunohistochemical studies for TSPO in healthy human brain revealed that TSPO is expressed in glial cells (microglia and astrocytes), oligodendrocytes and vascular endothelial cells [62, 63]. In post-mortem human AD brain samples, TSPO expression was demonstrated in microglia, macrophages and astrocytes but also in cells surrounding Aβ plaques [62] and only marginally significant differences in TSPO protein expression were found in temporal and frontal cortex between AD patients and healthy controls [63]. Furthermore, the TSPO burden did not correlate with neuropathology or with the glial response (microglia and astrocytes). In active and chronic active MS lesions, a strong correlation was shown between radioligand binding [(R)-[3H]PK11195 and [3H]PBR28], but astrocytic TSPO accounted for up to 65% of TSPO-positive cells in these lesions [64]. In PD rodent models, α-syn aggregates have been linked to microglial activation and correlated with expression of TSPO, but in PD patients TSPO PET has thus far yielded inconclusive results (recently reviewed by [65]).
Moreover, TSPO expression was also shown not to be phenotype-specific in microglia in active and chronic active MS lesions [64]. To directly correlate tracer binding to protein expression and distinguish cell types responsible for the radioligand uptake, Tournier et al. coupled fluorescence-activated cell sorting (FACS) to readout in radioligand-treated tissues (RTT) [66]. Using FACS-RTT, cellular origin of TSPO expression was shown to be highly dependent upon neuroinflammation type [66], and thus should be investigated for every pathology in order to attribute correct conclusions regarding TSPO PET imaging in human disease.
In line with these recent findings regarding TSPO protein expression, preclinical and clinical research results using TSPO PET in neurological diseases show highly variable outcomes. Nevertheless, some interesting recent imaging studies are outlined here. In a transgenic rat model of AD, using second-generation ligand [18F]FEPPA, researchers were unable to detect activated microglia that were MHC-II-positive. Similarly, in a rat model of stroke, TSPO PET signal was only found in the insult region, but not in remotely present MHC-II-positive microglia [67].
In healthy humans, a significant correlation between volume of distribution (VT) and age was found using second-generation ligand [11C]PBR28 [59]. Interestingly, another study showed that the binding potential (BPND) values of second-generation ligand [11C]DPA713 but not those of (R)-[11C]PK11195 increased with age, although [11C]DPA713 BPND values also did not reach significance [68]. Similarly, increased BPND values for [11C]DPA713 were found in AD patients in all ROIs, but for (R)-[11C]PK11195 BPND values were only elevated in precuneus [68], although patient cohorts for each tracer were different, so no direct comparison can be made.
(R)-[11C]PK11195 was used in a cohort of MS patients to follow-up on disease progress over 4 years [69]. Tracer uptake in normal-appearing white matter (NAWM) and thalamus of MS patients was significantly higher compared with healthy controls at the baseline scan, but no difference in cortical gray matter was observed. Interestingly, this is the first clinical study, to our knowledge, showing that higher TSPO binding predicts greater clinical disability, independent of relapsing status [69].
Taken together, being the ‘gold standard’ in PET imaging of neuroinflammation in the clinic, TSPO PET has had some success in revealing neuroinflammatory processes in human aging and disease. However, due to TSPO expression being non-specific for either pro- or anti-inflammatory status and even non-specific for a single cell type, there is still a need for the evaluation of other, more specific biological targets for PET imaging of neuroinflammation in human disease.
Emerging targets and PET tracers for imaging of activated microgliaTo overcome the limitations of TSPO PET, other neuroinflammation and microglia imaging targets are being explored (recently reviewed in Narayanaswami et al. [70]). In the next part of this review, we describe the emerging targets for PET imaging of microglia and the most recent preclinical and clinical results. Focus lies in the most promising targets for which significant advances were made with regard to target characterization and for which PET tracer development includes initial biological evaluation (Table 2).
TABLE 2. Activated microglia targets and their association with microglia phenotypes and potential tracers for PET imaging Molecular target Cellular localization Pro-/anti-inflammatory expression Tracers In vivo (pre-)clinical research stage References P2X7R Microglia (mainly), to a lower extend on oligodendrocytes and astrocytes Potentially specific to pro-inflammatory phenotype [11C]GSK1482160 First-in-human results in healthy volunteers warrant further clinical evaluation [80-82] [11C]SMW139 First-in-human results in healthy volunteers and MS patients warrant further clinical evaluation [75, 83] [11C]JNJ-54173717 No differences in uptake between healthy volunteers and PD or ALS patients [84-86, 143] [18F]JNJ-64413739 High inter-individual signal variability in non-human primates and human [87-89] P2Y12R Microglia Potentially specific to anti-inflammatory phenotype [11C]5 No in vivo data [25, 94] CB2R Microglia, astrocytes, neurons, endothelial cells No data on microglia phenotype expression [11C]NE40 Lower uptake in AD patients than healthy volunteers [
留言 (0)