
記住我
The Bacillus cereus sensu lato (s. l) also referred as B. cereus group, is a complex cluster of Gram-positive, spore-forming, rod-shaped bacteria comprising both pathogenic and non-pathogenic species (Okinaka and Keim, 2016). Over 18 members have been classified as part of the B. cereus group; B. albus, B. anthracis, B. cereus, B. cytotoxicus, B. luti, B. mobilis, B. mycoides, B. nitratireducens B. pacificus, B. paranthracis, B. paramycoides, B. proteolyticus, B. pseudomycoides, B. toyonensis, B. thuringiensis, B. tropicus, B. wiedmannii, and B. weihenstephanensis (Carroll et al., 2020a,b). The most prominent pathogenic species include B. anthracis, B. cereus, and B. thuringiensis, each displaying unique phenotypic and virulence traits. For instance, B. anthracis presents as non-motile, encapsulated, non-hemolytic in sheep blood agar, and sensitive to both γ-phage and penicillin (WHO, 2008), while B. cereus and B. thuringiensis are hemolytic, motile, non-encapsulated, and exhibits γ-phage and penicillin resistance (Vilas-Bôas et al., 2007; Kolstø et al., 2009).
Bacillus anthracis is a zoonotic pathogen which causes anthrax disease (Turnbull, 2002). The disease primarily affects livestock, wildlife and humans and may present as cutaneous anthrax, acquired through contact with contaminated food or meat of animal with the disease, and inhalation anthrax, from breathing in airborne anthrax spores (WHO, 2008). Pathogenicity in B. anthracis is primarily attributed to plasmids pXO1 and pXO2 which harbor the anthrax toxin genes that include protective antigen (pagA), lethal factor (lef) and edema factor (cya) and the poly-γ-D-glutamic acid (PGA) capsule genes (capABCDE), respectively (Turnbull, 2002; Pena-Gonzalez et al., 2018). In contrast, Bacillus cereus acts as an opportunistic pathogen capable of causing gastrointestinal and non-gastrointestinal infections (Kotiranta et al., 2000; Bottone, 2010). Gastrointestinal disease may manifest as diarrheal (linked to toxins such as hemolysin BL, non-hemolytic enterotoxin, and cytotoxin K) or emetic illness (due to the cereulide toxin encoded by cesABCDPTH) (Schoeni and Wong, 2005; Owusu-Kwarteng et al., 2017; Ehling-Schulz et al., 2006). On the other hand, Bacillus thuringiensis, often employed as a natural insecticide which is not harmful to humans, contains insecticidal crystal protein genes (cry and/or cyt), which may lead to its misidentification as B. cereus in the absence of these genes (Kolstø et al., 2009).
Due to the clinical, agricultural, and economic importance of B. cereus s.l, especially the pathogenic species, substantial research has focused on their classification and taxonomy (Baldwin, 2020; Carroll et al., 2020a,b). The classification of these species was historically based on characteristics such as hemolytic activity, colony morphology, γ-phage and penicillin activity and the detection of virulence markers that were alleged to be species-specific (Rasko et al., 2005; Kamar et al., 2013; Ehling-Schulz et al., 2019). However, the emergence of exceptional genomes, such as B. thuringiensis isolates carrying cry genes that phylogenetically grouped with B. anthracis (Kolstø et al., 2009), along with atypical B. cereus and B. cereus biovar anthracis strains carrying the pBCXO1 and pBCXO2 plasmids, which contain anthrax virulence factors (Antonation et al., 2016; Baldwin, 2020), and the loss of virulence plasmids in B. anthracis isolates (Marston et al., 2005), suggested that the identification and classification of B. cereus group isolates should not rely solely on phenotypes and virulence factors (Baldwin, 2020; Brézillon et al., 2015). In some instances, molecular analysis targeting virulence determinants of B. anthracis revealed the presence of homologous PGA that synthesizes capsular genes (capABCDE) of B. anthracis in other Bacillus species (pgsABCDE) (Lekota et al., 2016; Lekota et al., 2018). Moreover, varying phenotypic characteristics by expressed bacterial isolates and/or limited genomic databases may have led to nomenclatural discrepancies observed in the B. cereus group (Afshinnekoo et al., 2015; Abdelli et al., 2023), which could have profound implications for clinical and public health settings (Carroll et al., 2020a,b; Carroll et al., 2022a,b,c). To date, the phenotypic characteristics (i.e., hemolysis activity, capsule presence/absence), plasmids and virulence factors still play a critical role in routine diagnosis and surveillance of potential disease outbreaks associated with pathogenic members of the B. cereus group (Mogaji et al., 2024).
Genetic techniques such as DNA–DNA hybridization (DDH) (Goris et al., 2007), multiple-locus variable-number tandem repeat analysis (MLVA) (Marston et al., 2006), amplified fragment length polymorphisms (Guinebretière et al., 2008), 16S RNA (Braun et al., 2021) and 23S RNA (Sacchi et al., 2002) were entirely instrumental methods used to differentiate members of the B. cereus group. The growing scheme of technology and rising novel species saw an increase in the application of more gene-specific target sequencing, such as single- and multi-locus sequence typing (SLST and MLST) methods, which became and remained some of the essential tools used for the identification of the B. cereus group members (Guinebretière et al., 2008). The pantoate-ß-alanine ligase (panC) emerged as a popular target locus used in SLST to assign members of the B. cereus group into seven distinct phylogenetic groups (group I-VII) based on sequence variations providing superior resolution compared to other traditional methods such the 16S RNA (Guinebretière et al., 2008). The seven-group assignment was later updated to an eight-group panC group assignment/genomospecies (Group I-VIII) with species names as representative names for some of the assigned groups (Carroll et al., 2022a,b,c). Group I contains species closely related to B. pseudomycoides. Group II (mosaicus/luti) includes certain strains that exhibit unique characteristics but are less commonly identified. Group III (mosaicus) is notably the most diverse group containing strains with emetic toxin production (cereulide), anthrax virulence genes production, and traditional B. anthracis, B. cereus and B. thuringiensis species closely related to traditional B. anthracis. Group IV (B. cereus sensu stricto) is a group that contains strains closely related to B. cereus sensu stricto, frequently identified in clinical and food-related sources. Bacillus thuringiensis species are also located in Group IV. Group V (B. toyonensis) contains strains that have been less frequently characterized but are part of the broader Bacillus cereus group. Group VI (B. mycoides/paramycoides) primarily consists of psychrotolerant species, indicating their ability to grow at lower temperatures (Liu et al., 2018). Group VII (B. cytotoxicus) contains specific strains with cytotoxic effects related to cytotoxin K-1 (Fagerlund et al., 2004) and Group VIII (B. mycoides), which contains species closely related to B. mycoides.
The study of Liu et al. (2015) applied an integrated approach utilizing molecular techniques, including digital DNA–DNA hybridization (dDDH) with a ≥70% similarity threshold (Goris et al., 2007), MLST, and 16S RNA analysis. This approach clustered 224 genomes of B. cereus s.l into 30 clusters. Notably, dDDH analysis revealed that 20 genomes initially classified as B. cereus or B. thuringiensis exhibited unique genomic characteristics and were subsequently identified as B. anthracis, referred to as ‘anomalous B. anthracis’ to distinguish them from traditional B. anthracis isolates (Liu et al., 2015). In a separate study, 2,231 B. cereus group genomes were analyzed using Fast ANI, which found that approximately 66.2% of the genomes belong to several genomospecies at an ANI threshold of 95% (Carroll et al., 2020a,b). Furthermore, additional anomalous B. cereus group isolates were identified, including isolates with phenotypic features typical of B. anthracis, B. cereus, and B. thuringiensis, which exhibited an ANI ≥ 95% with canonical B. anthracis, yet lacked anthrax-specific virulence genes. This work also proposed a nomenclatural framework which integrated virulence detection, MLST, ANI and panC group assignment for the B. cereus group to harmonize phenotypic and genomic classification, aiming to reduce misclassification and misinterpretation of species in clinical and industrial settings, ultimately addressing public health implications (Carroll et al., 2020a,b).
The estimated pan-genome of the B. cereus group encompasses approximately 60,000 genes, with around 600 core genes shared by 99% of analyzed strains (Bazinet, 2017). This estimate, however, is likely evolving with the classification of new members and the availability of additional genomes. Pan-genome analysis has been instrumental in distinguishing essential core and accessory genes, revealing the functional versatility of the B. cereus group and the unique adaptive capacities of individual strains (Kim et al., 2017). While pan-genome analysis illuminates the broader genetic landscape, providing insights into environmental adaptability and metabolic functionality, whole-genome single nucleotide polymorphism (wgSNP) analysis offers a high-resolution for delineating differentiating genetic diversity within closely related strains (Bogaerts et al., 2023). Single nucleotide polymorphisms are evolutionarily stable markers that contribute to elucidating deep phylogenetic relationships among global strains (Pearson et al., 2004; Girault et al., 2014; Lekota et al., 2024). Given the monomorphic nature of B. anthracis, wgSNP analysis has proven valuable in differentiating traditional B. anthracis strains from other closely related B. cereus group members.
This study investigated the genomic cohesion among “anomalous” B. cereus group isolates, prompted by the identification of an isolate with phenotypic features consistent with B. cereus yet a phylogenetic profile aligning with typical B. anthracis. A genome-based comparative approach was employed, encompassing pan-genomic and wgSNP analyses of previously reported “anomalous” B. cereus group strains from the studies of Liu et al. (2015) and Carroll et al. (2020a,b), as well as newly sequenced isolates displaying typical B. anthracis and B. cereus phenotypes. The isolates investigated in this study were obtained from archival animal blood smears collected in anthrax endemic regions of Kruger National Park, South Africa, as part of their anthrax outbreak surveillance program. The isolates in this study were initially screened for anthrax virulence markers (pagA, lef, and capB) and the chromosomal marker Ba-1, with phenotypic characteristics of the isolated recorded as detailed in Ochai et al. (2024). The collaborative work of the surveillance program is put in place to monitor anthrax outbreaks caused by traditional B. anthracis and/or any other potential isolates that may cause anthrax disease.
2 Materials and methods 2.1 Sample collection and screeningBacterial cultures of samples collected between 2012 and 2015 (Table 1), were isolated from blood smears obtained from animal carcasses from the anthrax endemic regions of Kruger National Park (KNP) as described by Ochai et al. (2024). Briefly, blood smears on microscope slides were sterilely scrapped into 1.5 mL centrifuge tubes. Two hundred microliters of phosphate-buffered saline (PBS; Thermo Scientific, MA, United States) was added into the tube and half of the aliquot was spread plated on 5% sheep blood agar and incubated overnight at 37°C. All isolates with different colony morphologies were treated as different isolates and were sub-cultured on 5% sheep blood agar to obtain pure colonies. All the isolates were primarily subjected to classical methods that included microscopy, morphology, motility, hemolysin activity, γ-phage and penicillin sensitivity tests as described by the World Health Organization (WHO, 2008). The isolates were screened for the presence/absence of anthrax-toxin markers (pagA, lef, capB) and the B. anthracis chromosomal marker Ba-1 using SYBR green as prescribed by WHO (2008) and Taqman probe qPCR-based method as prescribed by Zincke et al. (2020). This was done in order to investigate any other Bacillus isolates which may carry anthrax virulence genes. The results are summarized in Supplementary Table S1, extracted from the study of Ochai et al. (2024).
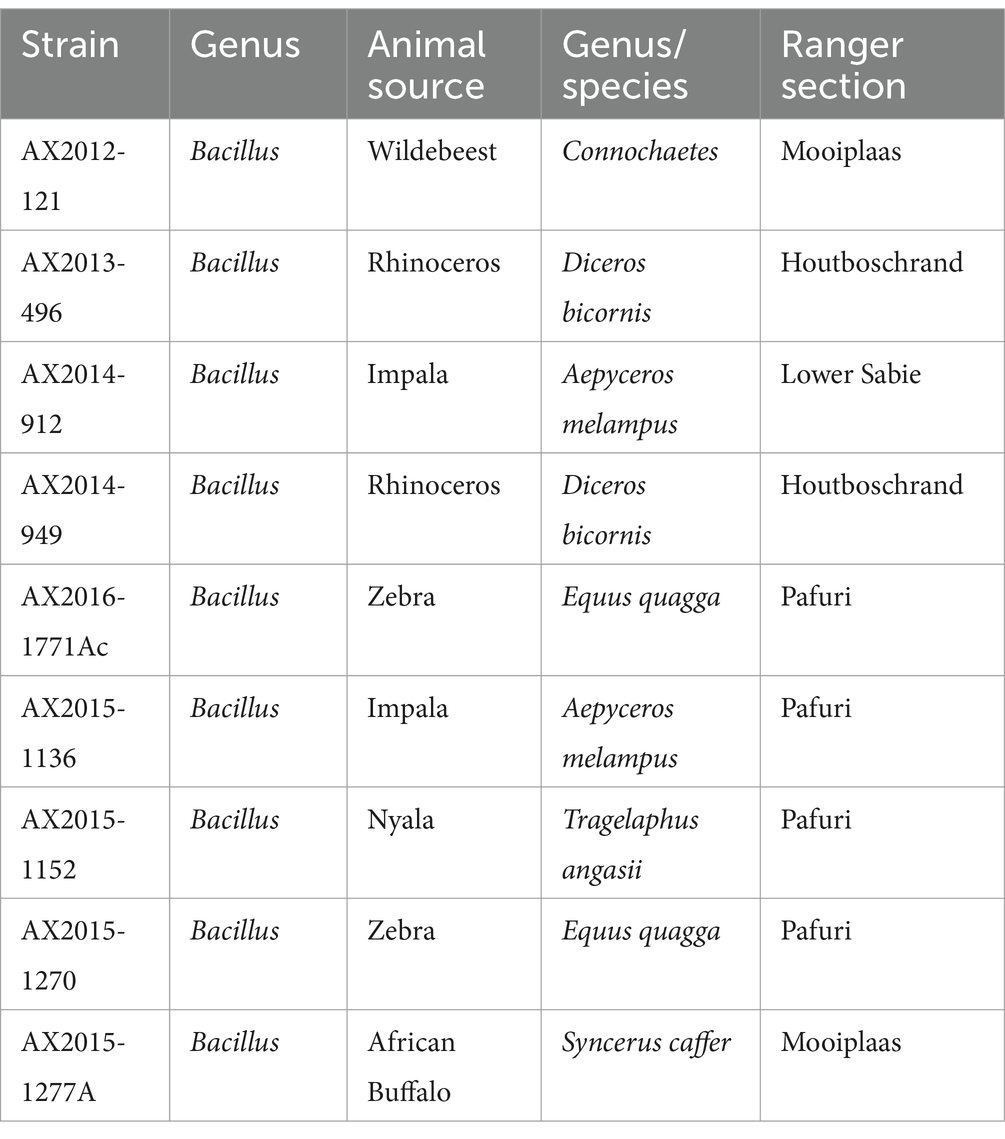
Table 1. Bacillus anthracis and B. cereus isolates from South Africa were cultured from blood smears from animal carcasses in the Kruger National Park, South Africa.
2.2 Genomic extractions and sequencingGenomic DNA of the isolates (n = 9) was extracted from overnight pure cultures using the Pure link Genomic DNA kit (Thermo Fisher Scientific, United States) following the manufacturer’s protocol. The concentration and quality of the DNA were determined using the Qubit 2.0 fluorometer (Thermofisher-Scientific, United States). The DNA was sent to the Agricultural Research Council-Biotechnology Platform (ARC-BTP) for whole genome sequencing. Sequence libraries of the isolates were constructed using the MGIEasy FS DNA Prep Kit (BGI, China) according to the manufacturer’s protocol. The prepared libraries were sequenced using the BGI MGISEQ-2000 platform (BGI Shenzhen, China) using the paired-end 2 × 150 bp to generate reads. Four genomic sequences (AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A) were described and published in our previous study (Magome et al., 2024). For this study, we included these four isolates as they form part of an anthrax surveillance study in KNP from the same culture collection of the B. cereus group.
2.3 Genome assembly and annotationDe novo sequencing assembly, adapter trimming and polishing were performed using Shovill v4.6.0 (Seemann et al., 2020). Transeq from EMBOSS was used for the translation of nucleotide sequences into amino acids (Rice et al., 2000). Diamond software was used to compare the contig sequences against the protein databases by BlastX (the following command option was used: diamond blastx --max-target-seqs 0 --more-sensitive --id 70 -p 8 --subject-cover 90) (Buchfink et al., 2015; Buchfink et al., 2023). In-house AWK, Python and Excel scripts were further used to filter the resulting data. The annotation of the selected isolates was first performed with Prokka v1.14 using the command option: prokka --cpus 8 --gcode 11 --rnammer --compliant --center XXX (Seemann, 2014), an updated annotation tool Bakta (Schwengers et al., 2021) was then used with the following command argument: bakta --db db --verbose --threads 8. The resulting output from annotations was further used as input for Roary 3.13.0 (the following options have been used: “-g 80000 -e --mafft -p 8 -r -qc -r -z -f”) (Page et al., 2015). Gene ontology classification of the pangenome data, was performed using DeepNOG (Feldbauer et al., 2020) and COG Classifier tools (Shimoyama, 2022). Whole genome SNP analysis was performed using Snippy v4.6.0 (Seemann, 2015). The clean alignment of the sequences was subjected to bootstrap =100 phylogenetic tree built with RaXML (Stamatakis, 2014).
2.4 Genome identification and phylogenetic placementThe sequenced genomes were first identified using the Pub-MLST species-ID search tool (Jolley et al., 2018) and the Genome Taxonomy Database (GTDB) v1.7.0 which incorporates the Fast Average Nucleotide Identity (ANI) on KBase app (Arkin et al., 2018). Pan-genomic placement together with ANI comparison using the sequenced genomes (n = 9) and the 18 recognized members of the B. cereus group obtained from GenBank was constructed using the integrated prokaryotes genome and pan-genome analysis web services (IPGA) v1.09 (Liu et al., 2022). A comprehensive comparative genomics analysis was conducted on 88 anomalous genomes of the B. cereus group, which were retrieved from GenBank and classified into three major species: B. anthracis (n = 36), B. cereus (n = 37), and B. thuringiensis (n = 15). This analysis included five B. cereus biovar anthracis strains (CAM, CAR, CI, DRC, and UFBc0001) sourced from animal samples in Cameroon, the Central African Republic, Côte d’Ivoire, and the Democratic Republic of the Congo. These strains have reportedly caused anthrax-like diseases in their hosts (Antonation et al., 2016; Baldwin, 2020). Additionally, the atypical B. cereus strain BC-AK, isolated from a kangaroo in China, was included, along with nine South African isolates from this study (Supplementary Tables S2, S3). In total, this analysis examined 103 genomes. Moreover, further description of the genomes was conducted using BTyper3 (Carroll et al., 2020a,b) that involved panC group assignment, PubMLST genome identification, GTDB identification and ANI comparison within the IPGA (Liu et al., 2022). The pan-genome trees were based on gene presence/absence and were visualized using tvBOT incorporated in the web-based tool ChiPlot (Xie et al., 2023).
2.5 Identification of mobile elements, virulence factors, and resistance genesVarious nucleotide/protein sequence databases, such as comprehensive antibiotic resistance database (CARD) (Alcock et al., 2023), ResFinder (Feldgarden et al., 2019), BacMet (Pal et al., 2014), BacAnt (Hua et al., 2021), MobileElementFinder (Johansson et al., 2021), PAIDB (Yoon et al., 2015), Iceberg2 (Liu M. et al., 2019; Liu B. et al., 2019) were used for mobile genetic elements and resistance gene predictions. Specifically, antibiotic resistance determinants were identified in each assembled genome using the ResFinder [−db ResFinder] (Feldgarden et al., 2019) with the minimum identity and coverage thresholds of 75 (−minid 75) and 50% (−mincov 50), respectively. Virulence factors in the sequenced genomes were mined using the Virulence Factor Database [−db vfdb] (Chen et al., 2016; Liu M. et al., 2019; Liu B. et al., 2019), using minimum identity and coverage thresholds of 75 (−minid 75) and 50% (−mincov 50), respectively. Further mining and annotation of antibiotic resistance genes (ARG), integrons and transposable elements was conducted using the BacAnt web-based tool (Hua et al., 2021), whereas BacMet was used to mine for metal resistance genes (Pal et al., 2014).
3 Results 3.1 Phenotypic and molecular characteristicsMicroscopic examination of the nine bacterial isolates revealed that all were Gram-positive rod-shaped cells. Notably, four isolates (AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A) displayed long rod chains, while the remaining five isolates (AX2012-121, AX2013-496, AX2014-912, AX2014-949, and AX2016-1771Ac) were characterized by shorter rod-shaped cells, presumptively identified as B. cereus. Among these, the latter five were β-hemolytic, motile, and showed γ-phage and penicillin resistance. Conversely, the four isolates previously identified as B. anthracis (AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A) were non-hemolytic, γ-phage and penicillin sensitive (Ochai et al., 2024). Molecular screening of these isolates for anthrax virulence genes indicated the presence of pagA, lef, and the chromosomal marker Ba-1 in B. anthracis isolates. The other five isolates, AX2012-121, AX2013-496, AX2014-912, AX2014-949, and AX2016-1771Ac, tested positive for the lef gene. The B. cereus isolate AX2014-912 tested positive for anthrax chromosomal marker Ba-1, while isolate AX2016-1771Ac amplified for the capB, lef, and pagA marker (Supplementary Table S1).
3.2 Genome metrics and identification of the Bacillus cereus group isolatesThe genome features of the sequenced isolates are presented in Table 2. Quality assessment of the assembled genomes showed an average of 99.43% completeness for all the genomes (n = 9). The genomes were initially identified using Pub-MLST and GTDB-tk v1.7.0, which incorporates Fast average nucleotide identity (ANI) by matching the query genomes against the closest reference strains incorporated in the database. The isolates AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A were identified as B. anthracis based on PubMLST species identifier and shared an ANI of ≥99% score when matched with the B. anthracis Vollum reference isolate (GCA_000007825.1) based on GTDB-tk v1.7.0. The isolates AX2012-121, AX2013-496, AX2014-912, and AX2014-949 were classified as B. cereus, based on PubMLST species identifier and each shared ≥98 ANI score when matched with B. cereus ATCC 14579 (GCA_000008725.1) using GTDB-tk v1.7.0. The genome size of the B. cereus genomes ranged from 5.34 Mb to 5.52 Mb, with a GC content ranging from 35.0 to 35.2%. Isolate AX2016-1771Ac was identified as B. cereus on the PubMLST species identifier. However, based on GTDB-tk v1.7.0, the closest reference genome to isolate AX2016-1771Ac was B. anthracis Vollum (GCA_000007825.1). The two genomes shared ≥97 ANI, which is well above the 95% ANI threshold typically used for prokaryotic species delineation (Jain et al., 2018).
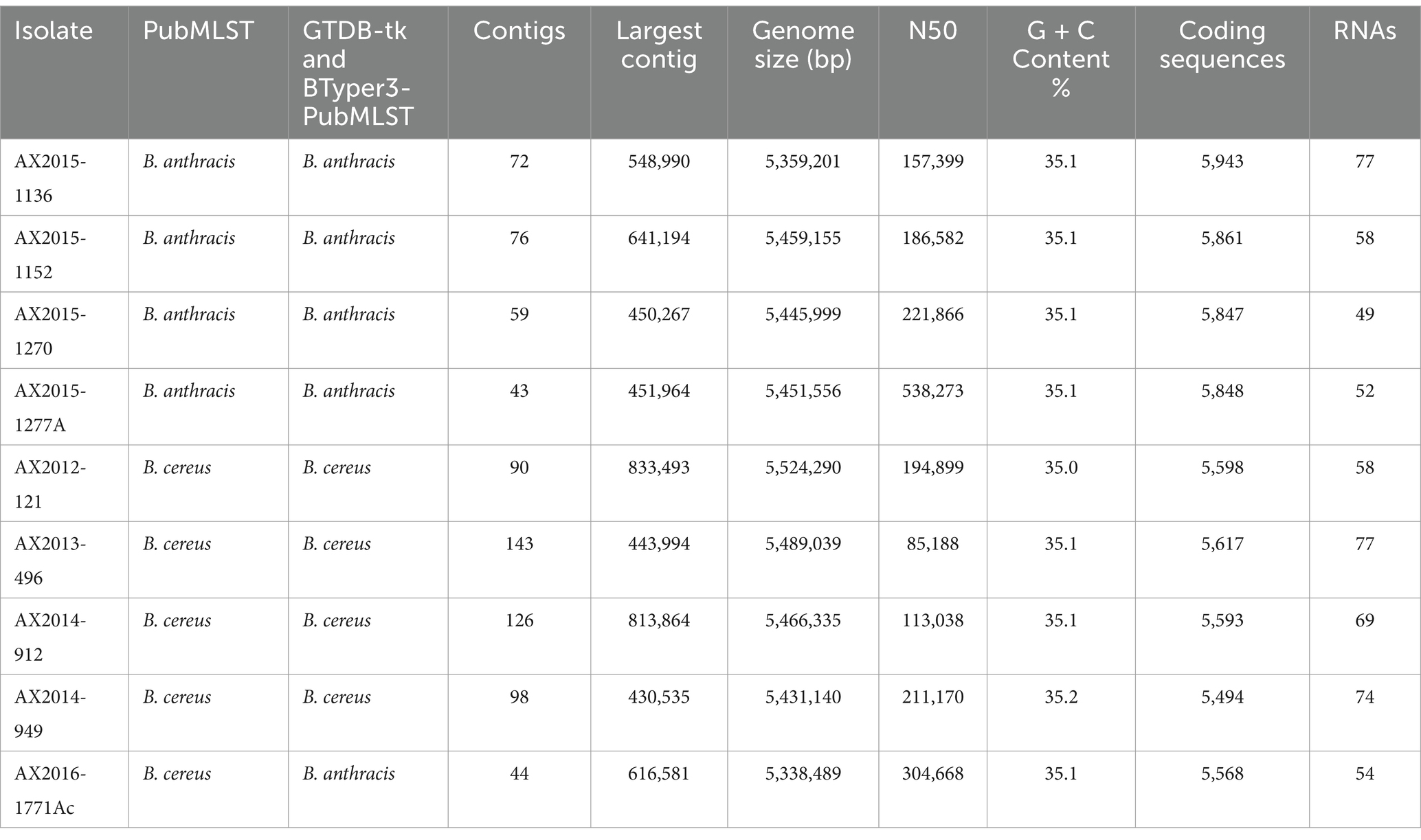
Table 2. Genome features of the nine South African sequenced Bacillus cereus group isolates.
The genome size of AX2016-1771Ac was 5.33 Mb with a GC content of 35.1% (Table 2). Phylogenetic placement using pan-genome and ANI analysis of the 18 recognized B. cereus group isolates in IPGA v1.09 (Figure 1) showed that isolates identified as B. cereus (AX2012-121, AX2013-496, AX2014-912, and AX2014-949) grouped with B. cereus ATCC 14579. The B. anthracis genomes (AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A) grouped more closely with B. anthracis Ames TYPE-STRAIN (GCA_000007845.1). However, isolate AX2016-1771Ac grouped closely with the B. anthracis isolates than with B. cereus isolates (Figure 1).
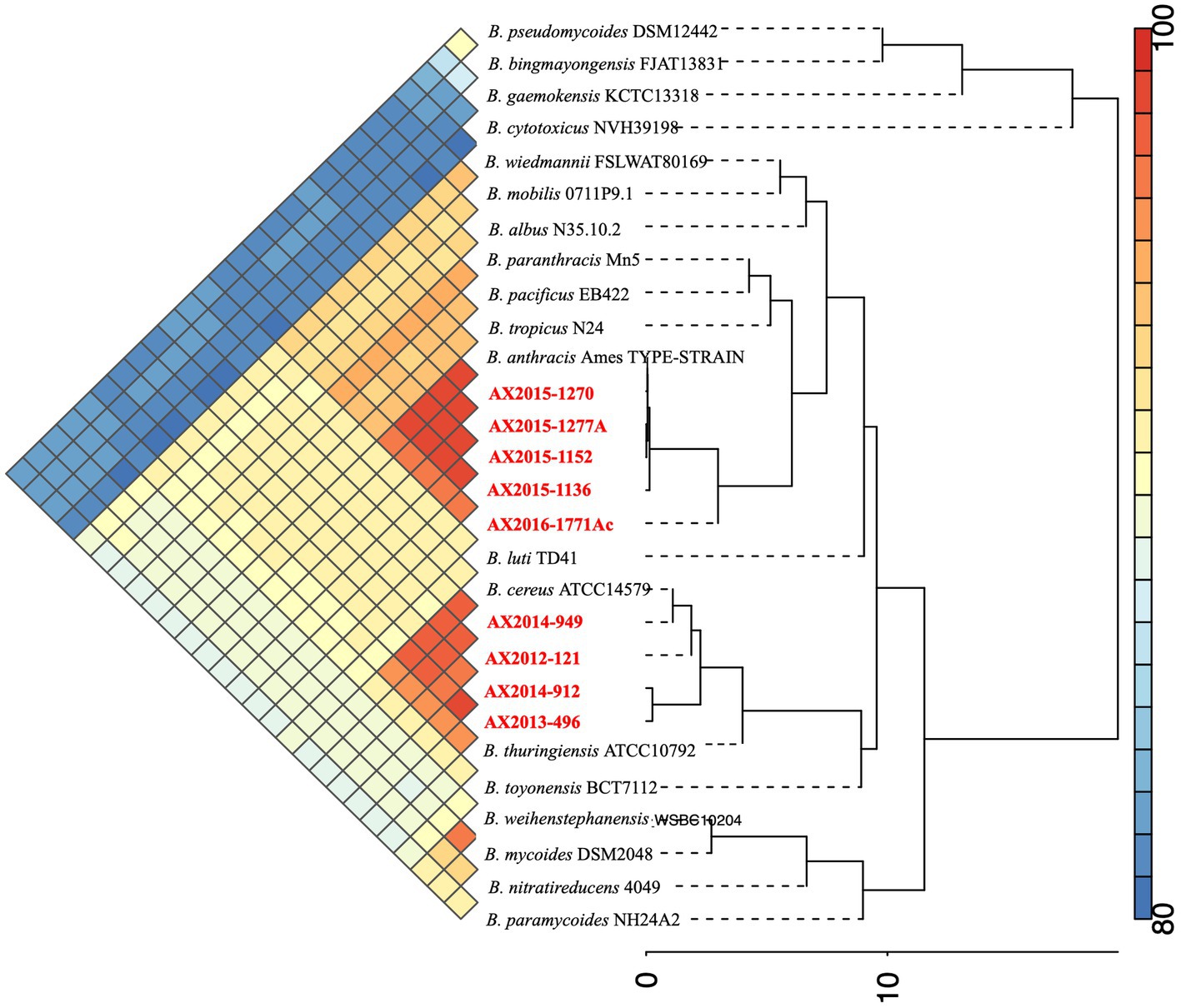
Figure 1. Phylogenetic placement and Average Nucleotide identity comparison using the 18 recognized members of the B. cereus group obtained from Carroll et al. (2020a,b), including the South African genomes (n = 9) from this study (marked in red).
3.3 Phylogenetic placement of the Bacillus cereus group genomes using various databasesThe pan-genome and wgSNP analysis revealed that four isolates (AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A) from South Africa identified as B. anthracis based on phenotypic characteristics, GTDB-tk and PubMLST classification, clustered with 26 additional genomes identified as B. anthracis on GenBank (PR01, PR02, PR05, PR06, PR07, PR08, PR09-1, PR09-4, PR10-4, Parent1, Parent2, Sterne, deltaSterne, BA_V770-NP1-R-ATCC 14185, Gmb1, Sen3, Sen2Col2, UT308, BAP417, BA781, Smith1013, Pasteur, A46, A1055, 2000031021, and 2000031052). This collective group, totalling 30 genomes, was consistently classified as B. anthracis based on GTDB-tk and BTyper3-PubMLST analysis and clustered in the mosaicus panC group III, sharing a ≥ 99% ANI (Figure 2; Supplementary Tables S2–S5). Given the genetic monomorphism characteristic of traditional B. anthracis strains, the wgSNP analysis (Figure 2B) supports the inclusion of these isolates within the B. anthracis lineage. Thus the 30 genomes are proposed to be accepted as part of the traditional or typical B. anthracis. This group of strains displayed diverse isolation sources, including laboratory/vaccine strains (n = 17), environment-soil (n = 1), animals (n = 9), and unknown source type (n = 3) from locations including the United States of America (USA), Senegal, Gambia, Pakistan and South Africa (Figure 2).
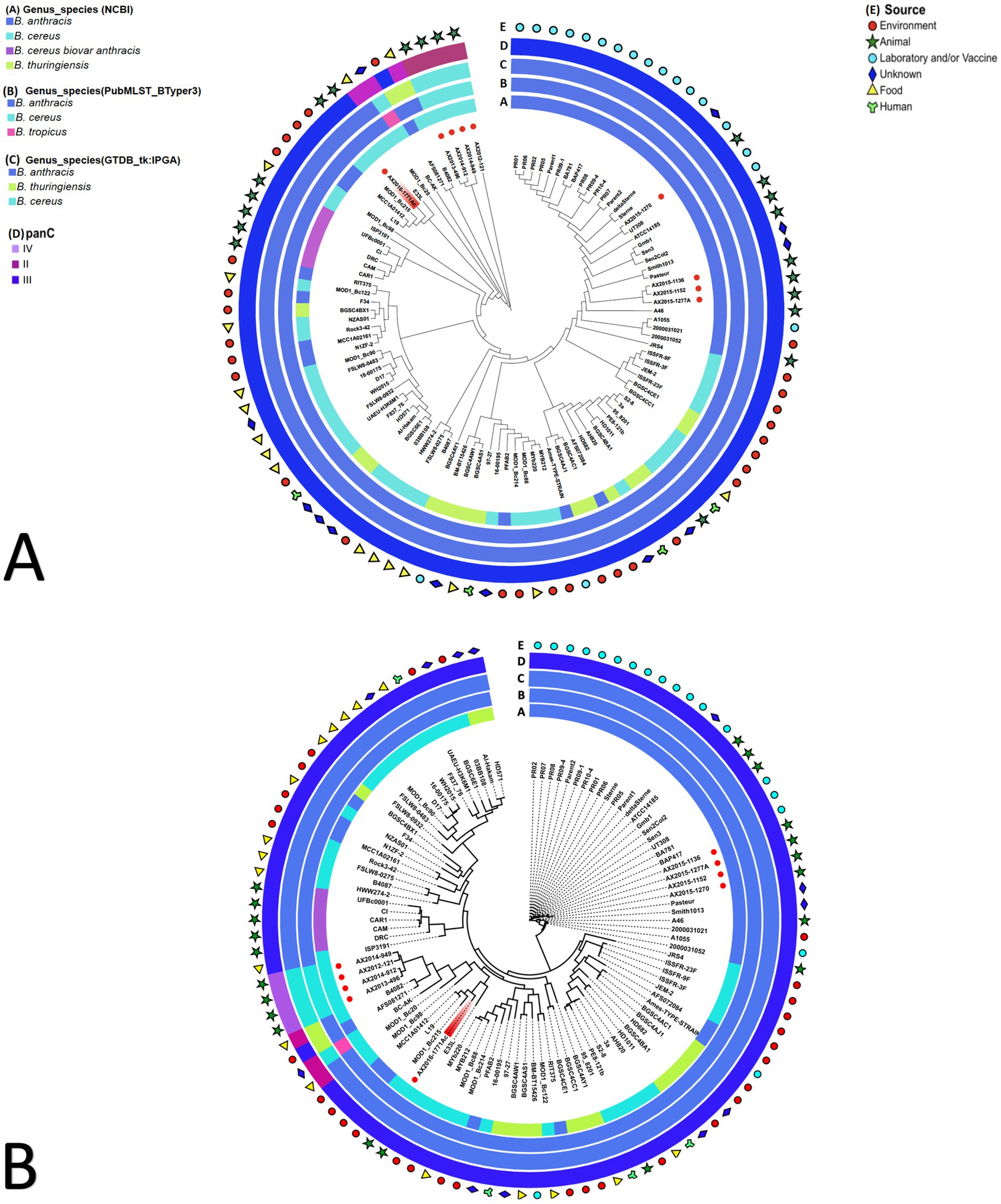
Figure 2. (A) Pan-genome and (B) whole genome single nucleotide polymorphism (wgSNP) analysis of 103 Bacillus cereus group genomes analyzed. The red dots in the inner circle indicate the South African isolates from this study. The AX2016-1771Ac highlighted in red. The rings surrounding the phylogenetic tree represent the following: (A) current genus species assignment as submitted on GenBank database; (B) BTyper3 PubMLST genome species assignment; (C) GTDB-tk-IPGA species assignment; (D) panC group assignment; (E) general source from which samples were obtained from.
Ten of the genomes classified on GenBank as B. anthracis (Ames TYPE-STRAIN, AFS072084, PFAB2, L19, MCC1A01412, AFS081271, MCC1A02161, N1ZF-2, RIT375, and F34) did not co-cluster with the previously mentioned 30 B. anthracis genomes based on pan-genome and wgSNP analyses (Figure 2). Furthermore, ANI analysis showed that the 10 genomes shared ≤98 ANI identity with typical B. anthracis genomes (AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A) (Supplementary Table S5). These genomes originated from diverse sources, such as hot spring (India, strain PFAB2), salt lake (Algeria, strain F34), sediment (South China sea: strains MCCC1A02161, MCC1A01412, N1ZF-2, and L19), soybean plant (strain AFS081271), corn plant (strain AFS072084), stem tissue of a Chamaecostus cuspidatus plant (Puerto Rico, strain RIT375) and laboratory/vaccine (Ames TYPE-STRAIN). Despite being classified as B. anthracis under BTyper3-PubMLST and as panC group III within the mosaicus group, one genome (AFS081271) was classified as B. thuringiensis by GTDB-tk in IPGA, aligning with isolate B4082 classified as B. cereus in GenBank isolated from a food source (pea soup in the Netherlands) (Figure 2). The genomes AFS081271 and B4082 were classified as B. thuringiensis based on GTDB-tk in IPGA v1.09 and classified as B. anthracis based on Btyper3 PubMLST belonging to panC group II of mosaicus/luti (strain B4082) and in panC group III of the mosaicus group (strain AFS081271) (Figure 2). The genomes AFS081271 and B4082 shared a ≥ 99% ANI when compared against each other, suggesting they are the same species. Moreover they clustered with four South African isolates (AX2012-121, AX2013-496, AX2014-912, and AX2014-949) identified as B. cereus under GTDB-tk and PubMLST (Figure 2).
The four B. cereus genomes, isolated from animal blood smears in South Africa, grouped under panC group IV in the B. cereus sensu stricto group based on Btyper3, showing 97–98% ANI among themselves and ≤ 91% ANI compared to the GenBank B. cereus genomes (Figure 2; Supplementary Tables S4, S5). Thirty-five GenBank B. cereus genomes were identified as panC group III of the mosaicus group and two GenBank B. cereus genomes (B4082 and MOD1_Bc20), isolated from food sources were identified as panC group II of the mosaicus/luti group, however they were classified as B. anthracis in BTyper3 (Figure 2). The results above highlight the discrepancies that exist within the various genomic identification databases which further complicate taxonomic classification. Additionally, an anomalous AX2016-1771Ac isolate, originating from a zebra blood smear, sequenced in this study, grouped with B. cereus (E33L and MOD1_Bc215) and B. anthracis (L19 and MCC1A01412) genomes using pan-genome and wgSNP analysis (Figure 2). These isolates, identified as B. anthracis under GTDB-tk and BTyper3-PubMLST, aligned with panC group III in the mosaicus group. The anomalous isolate AX2016-1771Ac shared 97–98% ANI with similar genomes from the South China Sea (strains L19 and MCC1A01412), United States baby wipes (MOD1_Bc215), and a Namibian zebra (strain E33L). Two B. thuringiensis strains (HD571 and Al-Hakam) that form part of the anomalous clustered with B. cereus strains BGSC6E1, 03BB108, F837_76, and UAEU-H3K6M1 as panC group III species of the mosaicus group (Figure 2). GenBank classified B. cereus strains (FLSW8-0483, WH2015, D17, and MOD1_Bc90) isolated from food in the United States and strain 16-00175 from France formed part of this group. Meanwhile, the B. anthracis Ames TYPE-STRAIN and AFS072084 genomes formed close associations with B. thuringiensis strains (BGSC4AJ1 from Mexico and BGSC4AC1 from India), with a notable ≥99% ANI score shared between B. anthracis Ames TYPE-STRAIN and B. thuringiensis BGSC4AJ1 (Supplementary Table S5).
The anomalous B. cereus ISP3191 genome isolated from food source, clustered closely with the B. cereus biovar anthracis strains (CAM, CAR, CI, DRC, and UFBc0001) isolated from animals (gorilla, chimpanzees, goat, and colobus monkey) in Western Africa, sharing ≥99% ANI. This cluster displayed a 97% ANI similarity with 82 other GenBank genomes, including the four typical B. anthracis strains (AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A) from this study. A subset of anomalous isolates (AFS081271, L19, B4082, MOD1_Bc20, MOD1_Bc98, and MOD1_Bc215) exhibited ≤96% ANI with B. cereus biovar anthracis isolates. Moreover, the B. cereus (AX2012-121, AX2013-496, AX2014-912, and AX2014-949) isolates from this study, shared <91% ANI with B. cereus biovar anthracis. The AX2015-1771Ac isolate shared ≥96% ANI score with B. cereus biovar anthracis isolates. However, AX2015-1771Ac shared <94% ANI with atypical B. cereus strain BC-AK. These results indicate that B. cereus group isolates may belong to different species clusters when a threshold ≥95% is adopted for all species in the same group.
3.4 Comparative annotation and functional analysis of Bacillus cereus group genomesTo predict gene content and perform pan-genomic analysis, 103 genomes within the Bacillus cereus group were annotated using two distinct tools: Prokka and Bakta. Initially, the annotation based on Prokka identified a total of 38,079 genes, with 26,173 (68. 73%) classified as hypothetical proteins. In contrast, Bakta annotation assigned 37,678 genes, only 5,391 (14.31%) were predicted to be hypothetical proteins. This suggests that Bakta may offer improved functional predictions for gene products within the B. cereus group, potentially reducing ambiguity around hypothetical proteins (Figure 3).
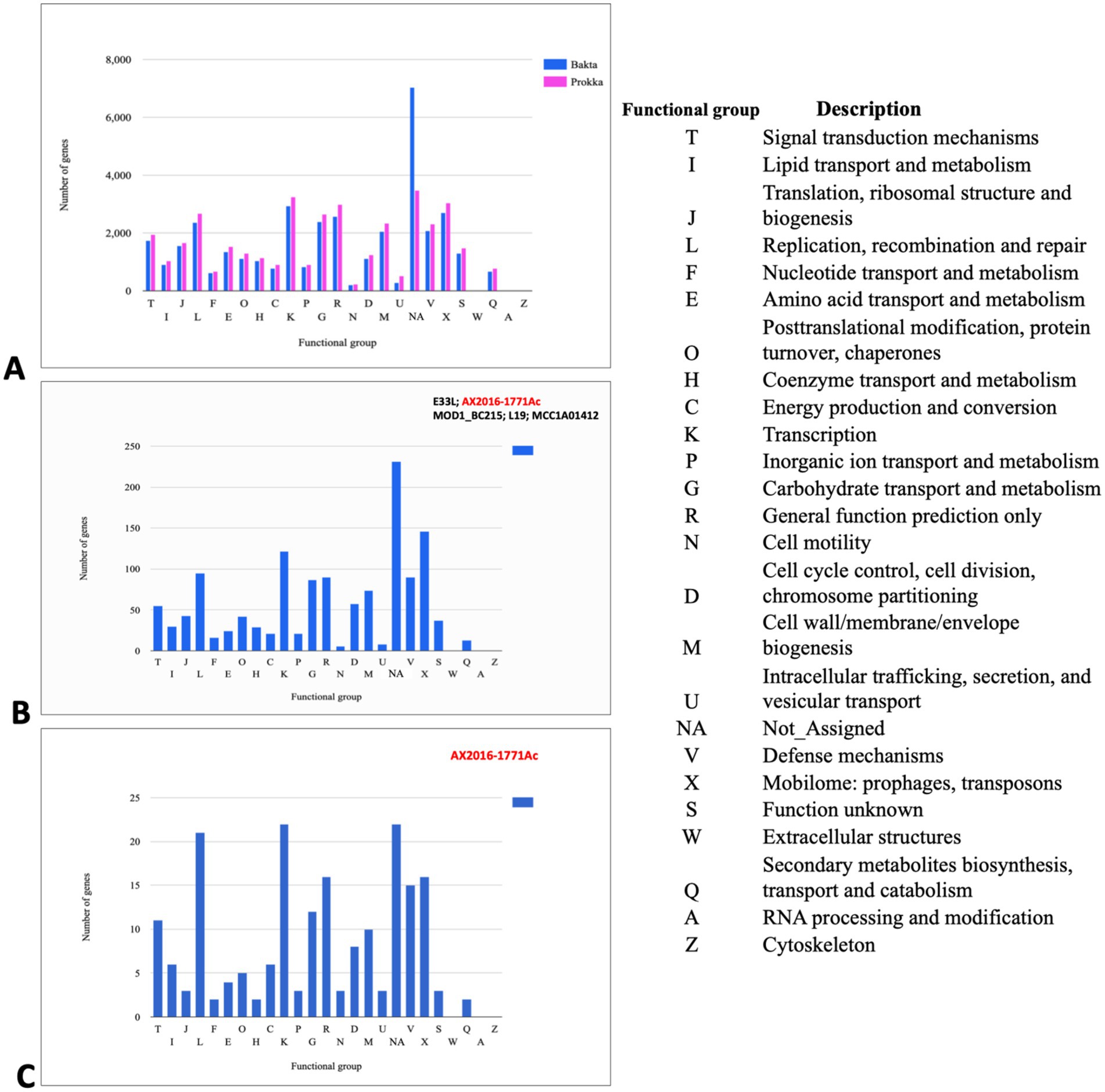
Figure 3. Cluster of orthologous groups (COGs) analysis indicating annotated genes in different functional groups (A) COG gene count comparison of the two annotation tools Prokka (pink) and Bakta (blue). (B) COG shared between the branch containing the five genomes B. cereus (AX2020-1771Ac, E33L, and MOD1_BC215) and B. anthracis (L19 and MCC1A01412) based on Bakta annotation. (C) COG corresponds to the set of genes that are unique to the anomalous sequenced isolate AX2016-1771Ac sequenced in this study. Each bar height corresponds to the total number of genes in the compartment that were assigned to the COG functional group. NA (Not Assigned compartment represents the genes that could not be assigned in a functional compartment).
In the Bakta annotation, 1,338 cloud genes were solely found in five anomalous isolates AX2016-1771Ac, B. anthracis strains (L19 and MCC1A01412) as well as B. cereus strains (E33L and MOD1_Bc215). Within this subset, 271 of these genes were assigned as hypothetical proteins. Additionally, 14 genes with distinct functional roles were identified exclusively in these five isolates. These genes included transport-related proteins (e.g., ABC transporter permease), structural proteins (e.g., spore coat protein), regulatory proteins (e.g., transcriptional regulators such as LysR and YdeE), metabolic enzymes (e.g., putative ubiquinone/menaquinone methyltransferase and NAD-dependent epimerase/dehydratase family protein), and other specific proteins (e.g., FAD-dependent oxidoreductase and DUF-domain proteins like DUF998, DUF952, DUF4430, and DUF3380). These unique genes may indicate specialized functionalities within these isolates. Functional categorization based on the cluster of orthologous groups (COG) analysis revealed that from the 1,338 clouds genes of the five compared strains, 146 genes (10.9%) were associated with mobilomes, including prophages and transposons functional group (X), followed by the transcription functional group (K) with 122 (9.1%) gene count, and the replication, recombination and repair functional group (L) with 95 (7.1%) gene counts (Figure 3B; Supplementary Table S6). Notably, the AX2016-1771Ac isolate contained 195 unique genes, which were not detected in other B. cereus group isolates, based on the COGs (Figure 3C).
3.5 Antibiotic resistance genes in Bacillus cereus groupA total of 16 antibiotic resistance genes (ARGs) were distributed across 103 Bacillus cereus group genomes analyzed in this study (Figure 4). The most abundant ARGs identified across all genomes (n = 103) were the beta-lactamase genes Bla1 (96%), BcII (95%), and Bla2 (78%). A distinct class A beta-lactamase gene, BcI, was exclusively detected in four B. cereus genomes as (AX2012-121, AX2013-496, AX2014-912, and AX2014-949) and the anomalous GenBank classified B. thuringiensis strain BM-BT15426. Additionally, the beta-lactamase gene, BlaTEM-116 was solely detected in the anomalous B. cereus strain FSLW8-0932, which was isolated from a food source in the United States. Another resistant gene, satA (encoding streptothricin N-acetyltransferase) was found in 48 genomes, but was absent in the B. cereus sensu stricto isolates (AX2012-121, AX2013-496, AX2014-912, and AX2014-949). The Fosfomycin resistance gene FosB2 was identified in 30 genomes, all suggested in this study to represent traditional/typical B. anthracis with high genomic similarity (≥99 ANI) (Figure 2). Notably, the 30 B. anthracis isolates, exhibited a consistent ARG profile that included BcII, Bla1, Bla2, FosB2, and satA (Figure 4), a typical profile for B. anthracis isolates.
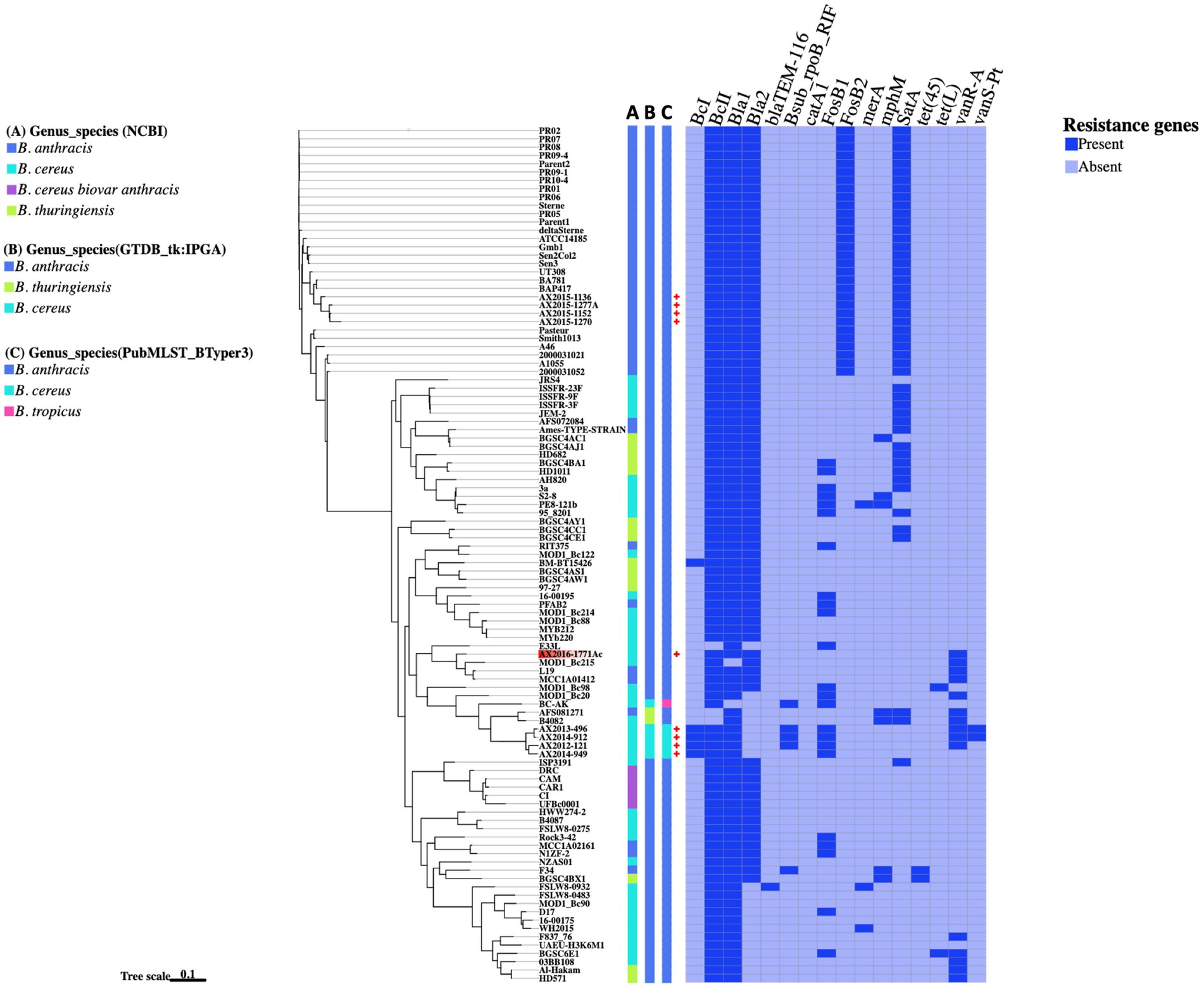
Figure 4. The presence and absence of resistance genes comparison between the Bacillus genomes. The phylogenetic tree is based on the wgSNP analysis of the 103 Bacillus genomes. The South African genomes from this study are marked with red crosses. A–C Blocks represents names assigned to the genomes on different genome identification databases: (A) Current genus species assigned to the genomes on NCBI; (B) Genus species assigned by the Genome taxonomy database in IPGA v1.09 (GTDB-tk); (C) Genus species assigned by the PubMSLT in BTyper3.
The fosfomycin gene FosB1 was detected in 23 anomalous genomes made up of 12 B. cereus isolates (16-00195, 3a, 95_8201, D17, E33L, MOD1_Bc20, MOD1_Bc214, MOD1_Bc98, PE8-121b, Rock3-42, S2-8, and BGSC6E1), four B. anthracis isolates (RIT375, PFAB2, NIZF-2, and MCC1A02161), two B. thuringiensis isolates (HD10111 and BGSC4BA1), including the atypical B. cereus BC-AK strain and the four B. cereus sensu stricto isolates (AX2012-121, AX2013-496, AX2014-912, and AX2014-949) from this study (Figure 4). Furthermore, the vancomycin resistance gene, vanR-A, was detected in 15 genomes that included B. cereus classified genomes (AX2012-121, AX2013-496, AX2014-912, AX2016-1771AC, 03BB108, B4082, F837_76, MOD1_Bc20, MOD1_Bc215, and BGSC6E1), B. anthracis genomes (AFS081271, MCC1A01412, and L19) and B. thuringiensis (HD571 and Al-Hakam). The gene rpoB known to confer resistance to rifampicin was detected in the South African B. cereus strains (AX2012-121, AX2013-496, and AX2014-912), the atypical B. cereus BC-AK strain associated with anthrax-like disease, as well as in the anomalous B. anthracis F34 strain isolated from Algeria. The vanS-Pt gene that is associated with ruminant microbiota in Paeniballicus strains (Guardabassi et al., 2005) and vancomycin resistance was only detected in two B. cereus genomes AX2013-496 and AX2014-912 sequenced in this study. The tetracycline efflux pump tet(45) was detected in anomalous B. anthracis F34 and B. thuringiensis BGSC4BX1 isolates. The tetracycline efflux pump tet(L) was detected in the anomalous B. cereus isolates BGSC6E1 and MOD1_Bc98. The sequenced typical B. cereus isolates AX2013-496 and AX2014-912 each presented seven ARGs namely the BcI, BcII, Bla1, rpoB, FosB1, vanR-A, and vanS-Pt. The B. cereus AX2012-121 strain contained six ARGs namely the BcI, BcII, Bla1, rpoB, FosB1, and vanR-A. The B. cereus AX2014-949 strain contained only four ARGs, which were identified as BcI, BcII, Bla1, and FosB1. The anomalous B. cereus strain AX2016-1771Ac also contained four ARGs namely BcII, Bla1, Bla2, and vanR-A. Bacillus cereus and B. thuringiensis isolates indicated a potential to carry varying resistance gene profiles from the typical B. anthracis isolates.
3.6 Virulence factors in Bacillus cereus groupIn this analysis, 117 virulence factor genes were found across 103 genomes analyzed in this study, notably genes which form part of a cluster were counted as individual genes. It has already been established that the 88 GenBank isolates B. anthracis (n = 36), B. cereus (n = 37), and B. thuringiensis (n = 15) do not contain the anthrax virulence genes: edema factor- cya, lethal factor- lef, the protective antigen -pagA located on the pXO1 plasmid (Carroll et al., 2020a,b). In this study, the virulence genes cya, lef and pagA were found present in the confirmed B. anthracis isolates AX2015-1136, AX2015-1152, AX2015-1270, and AX2015-1277A, atypical B. cereus BC-AK isolate and the B. cereus biovar anthracis isolates (UFBc0001, CAR, CAM, and CI). The anomalous isolate AX2016-1771Ac did not contain the anthrax virulence genes cya, lef and pagA. All the capsular genes cap-ABCDE and the capsule synthesis transcriptional regulator genes acpA and acpB were detected in the identified and classified traditional B. anthracis isolates (deltaSterne, AX2015-1152, AX2015-1270, AX2015-1277A, A1055, 2000031021, 2000013052, A46, Smith1013, and Pasteur), B. cereus biovar anthracis isolates (UFBc0001, CI, CAM, CAR, and DRC) and the B. cereus BC-AK isolate. The B. cereus 03BB108 isolated from the environment (dust particles) in the United States and the B. anthracis isolate N1ZF-2 from sediments in China both contained the capsule synthesis regulator gene acpB and the capsule genes capA and capC. The virulence regulator gene of B. anthracis (atxA), and the has-ACB gene cluster that encodes for the hyaluronic acid capsule was detected in sequenced B. anthracis isolates (AX2015-1136, AX2015-1152, AX2015-1270, AX2015-1277A), and anomalous B. anthracis strains BAP417 and BA781, the B. cereus BC-AK isolate including the B. cereus biovar anthracis isolates UFBc0001, CI, CAM, and CAR (Figure 5).
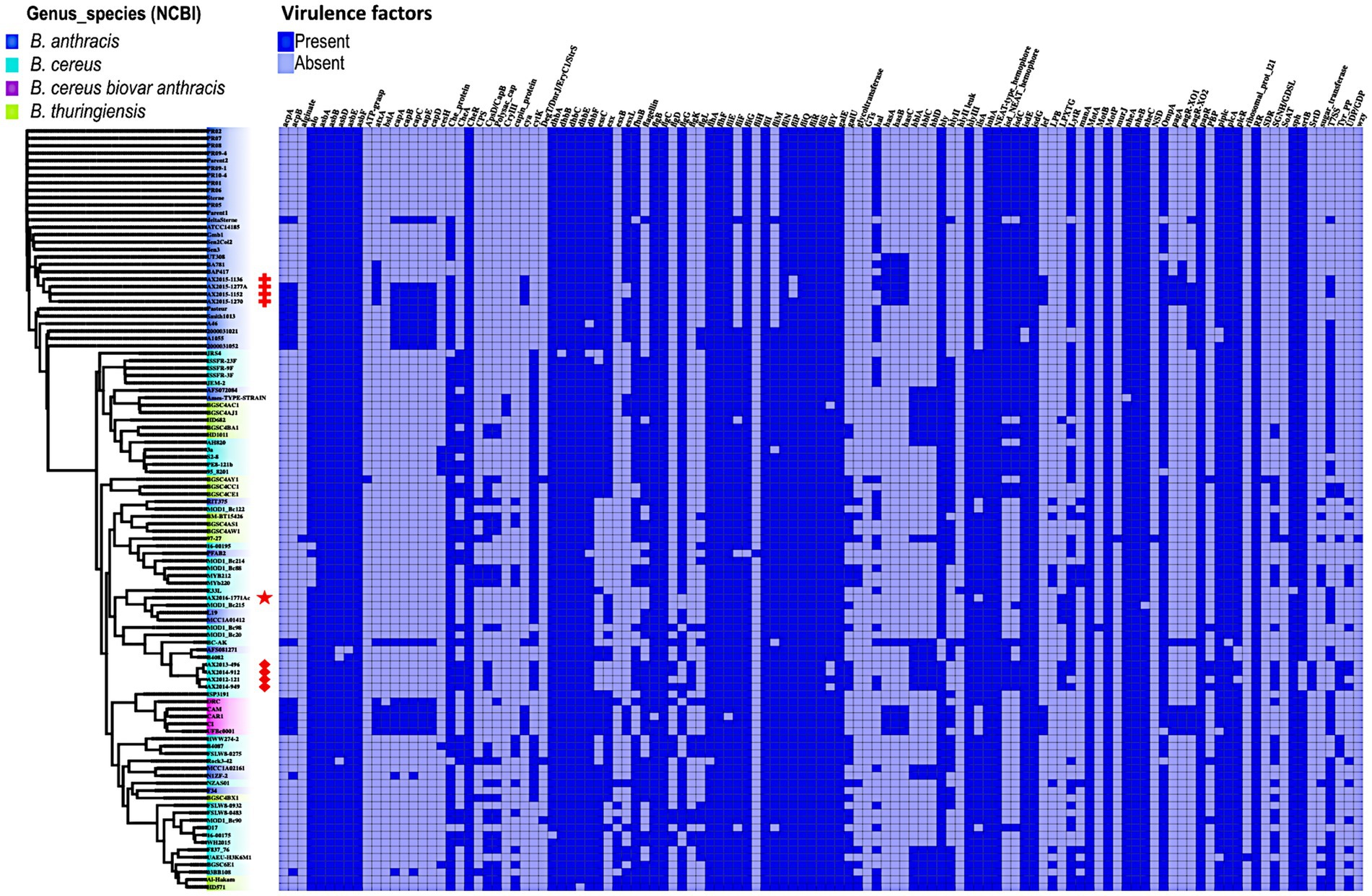
Figure 5. Presence and absence of virulence genes/factors identified in Bacillus genomes (n = 103) used in this study. The phylogenetic tree is based on the wgSNP of the Bacillus genomes. The South African isolates classified as B. anthracis (n = 4) are marked with red crosses; B. cereus s. s (n = 4) red diamonds shape, anomalous isolate AX2016-1771Ac marked with the red star.
The insecticidal crystalline delta-endotoxin gene cryIII was detected in the B. anthracis Ames TYPE-STRAIN and B. thuringiensis BGSC4AC1 and BGSC4AJ1 strains that shared a common ancestor. At least two or more genes belonging to the gene clusters asb-ABDEF encoding for the biosynthetic machinery for petrobactin and the dhb-ABCEF bacillibactin which are siderophores involved in iron acquisition of Bacillus species were detected in all the genomes. Although B. anthracis is non-motile, gene clusters related to flagellum synthesis (flg, flh, and fli) were present in all the genomes. The non-hemolytic enterotoxin nhe-ABC cluster was present in over 99% percent of the genomes. Whereas genes of the hemolytic enterotoxin gene complex hbl-ACD were detected in 24 genomes which included anomalous B. cereus, B. anthracis, and B. thuringiensis. The cytotoxin gene cytK was present in 49.6% of the genomes classified as B. anthracis, B. cereus, and B. thuringiensis, however our sequenced B. anthracis strains lack this gene. The hydrolase cesH gene which forms part of the cereulide operon was detected in six anomalous B. cereus strains (3a, 95_8201, BA087, BGSC6E1, PE8-121b, and S2-8). A more detailed description of the virulence genes is included in Supplementary Table S7.
3.7 Determination of the insertion sequences on the 103 genomesA total of 50 insertion sequences (IS) and five mobile insertion cassettes (MIC) were detected among the 103 genomes (Figure 6). The 50 IS were distributed as follows: IS231 (n = 15), ISBce (n = 15), ISBth (n = 16), ISBwe (n = 2), ISBt (1), ISBsp8 (n = 1), and IS232 (n = 1). Mobile insertion cassettes included MICBan1 (n = 1), MICBce (3) and MICBth (n = 1). The insertion sequence IS231L was the most prevalent, as it was detected in all 103 genomes analyzed in this study. Among the 30 genomes classified as B. anthracis in this study, insertion sequences IS231 (L and S), ISBt (2 and 7) and the insertion cassette MICBan1 were found in AX2015-1136, AX2015-1152, AX2015-1277A, AX1270, and BA781. Other insertion sequences, such as IS2321, ISBwe, and ISBt variants, were less frequent than ISBce and ISBth. The four South African B. anthracis isolates contained more IS than the B. anthracis isolates in the same cluster (Figure 6). The anomalous B. cereus strain AX2016-1771Ac strain sequenced in this study included the insertion sequences IS231L, ISBce (17 and 19), ISBth (4 and 7) and the insertion cassette MICBan1. None of the insertion genes can discriminate typical B. anthracis from the anomalous B. cereus group strains compared in this study. The B. anthracis Ames TYPE-STRAIN and B. thuringiensis BGSC4AJ1 and BGSC4AC1 contained similar insertion sequence profiles, which included IS231 (C, E, H, L), ISBce (3 and 15), ISbth (16 and 17), and MICBan1.
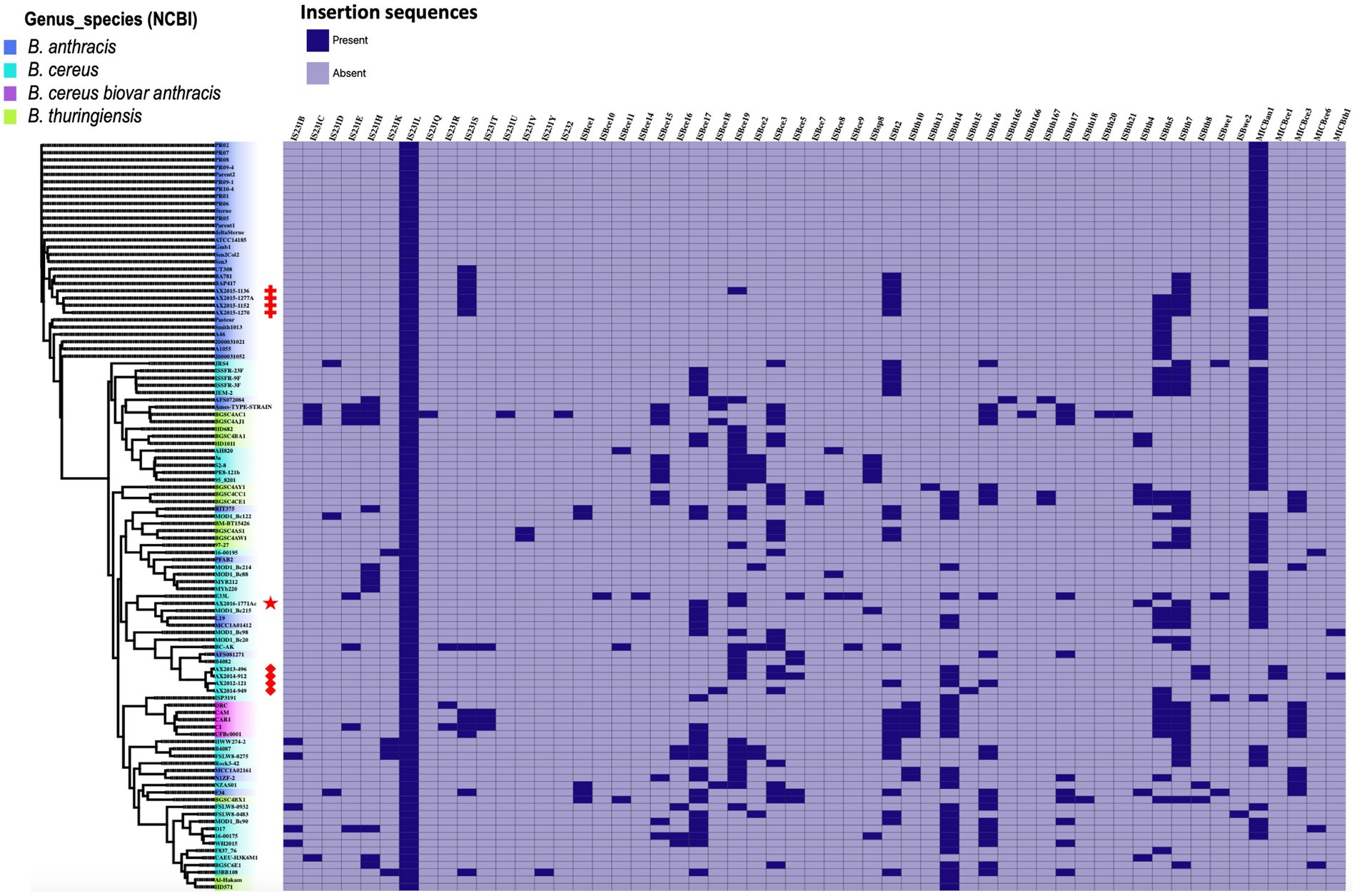
Figure 6. Heat map showing the presence and absence of insertion sequences and mobile insertion cassettes detected on 103 B. cereus group genomes analyzed in this study. The phylogenetic tree is based on the wgSNP analysis. The South African isolates classified as B. anthracis (n = 4) are marked with red crosses; B. cereus s. s (n = 4) red diamonds shape, anomalous isolate AX2016-1771Ac marked with a red star.
4 DiscussionIn this study, a comparative genomics approach was employed to analyze the nine B. cereus group isolates (Table 1), from animal blood smears that also included previously reported four B. anthracis strains from anthrax surveillance in KNP (Magome et al., 2024). Pan-genomic and wgSNP analysis was conducted to investigate previously reported “anomalous” B. cereus group strains (Liu et al., 2015; Carroll et al., 2020a,b), as well as newly sequenced isolates displaying typical B. anthracis and B. cereus phenotypes. The study highlights and supports some of the approaches used in the proposed framework by Carroll et al. (2020a,b). We further indicate through a restrictive ANI value of 99% and wgSNP analysis traditional B. anthracis can be distinguished from other closely related species which form part of the mosaicus group. Additionally, mobile genetic elements, including insertion sequences, virulence and antibiotic resistance genes across the genomes were investigated to contribute to the growing body isolates that form part of the B. cereus group.
The isolates in this study were initially screened for anthrax virulence markers (pagA, lef and capB) and the chromosomal marker Ba-1, with phenotypic characteristics of the isolated recorded as detailed in Ochai et al. (2024). An anomalous strain (AX2016-1771Ac) isolated from Equus quagga (Zebra) was discovered when it grouped phylogenetically with B. anthracis isolates Ochai et al. (2024). Notably, the strain was initially classified as B. cereus using classical microbiological tests, presenting as hemolytic, motile, and characterized as Gram-positive rods with short chains. Furthermore, together with other Bacillus isolates (Ochai et al., 2024), five B. cereus isolates sequenced in this study presented with positive qPCR detection for the anthrax virulence markers lef and/or pagA and/or chromosomal marker Ba-1 (Supplementary Table S1). The findings warranted further genomic investigations alongside reference atypical strains B. cereus and B. cereus biovar anthracis, which have reportedly caused anthrax-like diseases in humans and animals (Antonation et al., 2016; Baldwin, 2020).
While classical microbiology techniques, such as culturing and microscopy are still essential for bacterial identification, particularly in anthrax endemic regions (WHO, 2008; Ochai et al., 2024), molecular diagnostics often reveal discrepancies. The work of Ochai et al. (2024) showed that Peribacillus, Lactobacillus, and Priestia species were positive for anthrax virulence markers (pagA, lef, and cap). As an example, the PCR amplicons of pagA from Peribacillus, Lactobacillus, and Priestia isolates were sequenced and the BLASTn identification did not match with the pagA of B. anthracis (Ochai et al., 2024). Further analysis that involved genomic analysis and BLASTn comparison of the anthrax virulence markers in the Priestia and B. cereus group genomes revealed that these genes (pagA, lef, and capB) were only present in the four confirmed B. anthracis genomes with 99.9–100% alignment (Magome et al., 2024). In this study, several isolates with B. cereus-like phenotypes were positive for the anthrax-specific lef gene in qPCR assays but did not indicate lef gene in the genome after sequencing. The lef gene was found in the following isolates, with 99.9–100% in the four B. anthracis isolates, B. cereus biovar anthracis isolates and the atypical B. cereus strain. This is to be expected as anthrax virulence factors have been detected in these strains and were implicated in causing the anthrax disease to their host animal (Antonation et al., 2016; Baldwin, 2020). Previous studies have reported on false positives occurrence for anthrax virulence markers in non-B. anthracis strains from blood smears in anthrax endemic region in Kruger National Park (Lekota et al., 2016; Lekota et al., 2018). As thus the risk of misidentification in routine diagnostics, which could potentially compromis
留言 (0)