
記住我
According to the World Health Organization (WHO) Global Tuberculosis Report 2024, an estimated 0.40 million individuals worldwide were affected by multidrug- or rifampicin-resistant tuberculosis (MDR/RR-TB) in 2023, with only 44% of cases enrolled in treatment programs (1). For the 2021 patient cohort (the most recent with available data), the global treatment success rate for MDR/RR-TB was 68%. These figures underscore the persistent challenge that MDR/RR-TB poses to global public health (1).
China reported an estimated 741,000 new tuberculosis (TB) cases in 2023, with an incidence rate of 52 per 100,000 population. While the country’s TB incidence ranks at the lower-middle level globally, it remains the third-largest contributor to the global TB burden, accounting for approximately 6.8% of total cases. The WHO classifies China as a high-burden country for TB, AIDS-related TB, and MDR/RR-TB (1).
Wuhan, a major transportation hub and the fourth-largest city in China with a resident population exceeding 10 million, is also the economic center of central China. It hosts the second-largest number of colleges and universities nationwide, contributing to a complex population structure that impacts TB transmission. In 2023, the TB notification rate in Wuhan closely mirrored the national average (43.65 vs. 43.49 per 100,000 population), as reported by the National Tuberculosis Program system (2).
Rapid and accurate detection of drug-resistant TB is essential for controlling the epidemic. While phenotypic drug susceptibility testing (DST) remains the gold standard, its diagnostic timeframe of 10 days to 2 months limits its effectiveness in rapid intervention. Molecular diagnostic tools, such as GeneXpert MTB/RIF, have significantly reduced diagnostic time but are restricted to detecting mutations at high-frequency loci with variable sensitivity and specificity across regions (3, 4). Whole-genome sequencing (WGS) offers a comprehensive solution by detecting all known mutations in drug resistance genes with high sensitivity and specificity, enabling significant reductions in diagnostic time (5, 6).
Beyond diagnostics, WGS has proven to be an effective tool for genotyping, cluster identification, and studying the transmission dynamics of TB (7, 8). While alternative genotyping methods such as insertion sequence 6,110-restriction fragment length polymorphism (IS6110-RFLP) (9), spoligotyping (10), and mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU-VNTR) analysis can provide insights (11), they are limited in scope and lack the accuracy offered by WGS. Consequently, the accuracy of these genotyping methods is limited compared to the comprehensive resolution offered by WGS (12).
Drug resistance mutations and clustering of Mycobacterium tuberculosis isolates are influenced by phylogenetic lineages, which can also be elucidated through WGS. To date, no studies have explored the genetic diversity and recent transmission dynamics of multidrug-resistant tuberculosis in Wuhan using WGS. Our previous research (13) utilized a combination of 24-locus MIRU-VNTR, deletion-targeted multiplex polymerase chain reaction (DTM-PCR), and sequencing of resistance-associated genes to genotype MDR-TB isolates from Wuhan Pulmonary Hospital in 2017. This study utilized WGS data to analyze the spectrum of drug resistance mutations, identify clusters, and examine transmission patterns in Wuhan.
Materials and methods Isolates from registered casesIn 2017, 341 MDR-TB patients were registered in the Wuhan TB reporting network, a component of the National Tuberculosis Program system. Of these, 151 cases were identified at Wuhan Pulmonary Hospital, one of two designated tuberculosis treatment centers in Wuhan. After excluding two isolates that were unavailable, a total of 149 isolates were included in this study and preserved in the strain library at −80°C using glycerol-containing 7H9 medium. These 149 subcultures were subjected to WGS after their initial isolation.
DSTDST was performed using the proportion method on the Lowenstein-Jensen medium (Baso Company, China). Susceptibility to a total of eleven anti-tuberculosis (anti-TB) drugs, comprising five first-line and six second-line drugs, was assessed in accordance with WHO guidelines (14). The H37Rv strain was used as a quality control reference throughout the testing process.
WGSWGS of 149 MDR-TB clinical isolates was outsourced to Shanghai Jingnuo Biotechnology Co., Ltd., which provided the raw sequencing data. Sequencing was performed using the Illumina NovaSeq 6,000 platform (Illumina, United States) with paired-end sequencing (PE150). The bioinformatics pipeline used for analyzing raw sequencing reads, including the identification of single nucleotide polymorphisms (SNPs) and drug resistance gene mutations, is outlined in Figure 1.

Figure 1. Flowchart depicting the bioinformatics data analysis pipeline for identification of SNPs and drug resistance gene mutations from raw sequencing reads.
WGS data collation and cleaningThe WGS data collation and cleaning process involved the following steps: Raw sequencing reads from 149 strains were processed using Fastp (version 0.20.0) to automatically remove reads containing adaptor sequences or those of low quality. High-quality reads were then aligned to the reference genome of the standard Mycobacterium tuberculosis strain H37Rv (GenBank accession: NC_000962.3), obtained from the NIH GenBank database. Alignment was performed using BWA software (version 0.7.17), and sequencing depth and genome coverage were calculated. Next, sequencing data were filtered based on specific thresholds: sequencing depth > 100× and genome coverage >95%. From the processed data, 149 datasets met these criteria, with an average sequencing depth of 205.9× and genome coverage of 99.29%. These datasets were deemed suitable for further analysis.
Identification of single nucleotide polymorphismsSNPs were identified by aligning the sequencing data against the reference genome Mycobacterium tuberculosis H37Rv. SNP calling was conducted using Freebayes (version 1.3.2).
To ensure data quality, candidate SNPs were filtered using Bcftools (version 1.7). Filters excluded SNPs with sequencing depth below 10 or mapping quality scores less than 100. As a result, a refined set of high-confidence SNPs was obtained for downstream analysis.
Drug resistance gene mutation detectionWhole-genome SNPs identified in this study were compared against a database of known drug-resistance mutations. This comparison facilitated the characterization of drug-resistance gene mutations in each strain. Additionally, these mutations were analyzed to predict the likelihood of drug resistance in the isolates.
Phylogeny constructionThe mutation matrix for each strain was generated using Snippy software (v4.4.3) based on identified mutation sites. From this mutation matrix, an SNP distance matrix was computed to determine the genetic relationships between strains. Strains with an SNP difference ≤ 12 were identified as clustered. A maximum-likelihood phylogenetic tree was then constructed using core SNPs via IQ-TREE software (v2.1.4).
Cluster definitionIn this study, genomic clusters were defined based on an SNP cutoff value of ≤12, as established in the majority of prior studies (15, 16). The minimum estimate of the recent infection rate was calculated using the formula (13) nc−c/n , where nc represents the total number of clustered isolates, c is the number of clusters, and n represents the total number of isolates analyzed.
Modern Beijing sublineage definitionThe Modern Beijing sublineage was identified by the presence of both the C1477596T (ogt12) mutation and a single copy of IS6110 in the NTF region (17).
Statistical analysisStatistical analyses were conducted using SPSS version 21.0 (SPSS, United States). The Pearson chi-square test or Fisher’s exact probability test was applied to assess the statistical significance of differences in rates (e.g., mutation frequency) between groups, with a significance level of α = 0.05. Gene mutation frequencies below 3% were excluded from the analysis tables to ensure robust data representation.
Ethics statementEthical approval for this study was granted by the Wuhan Pulmonary Hospital Medical Ethics Committee (protocol number: 2018-12). Written informed consent was obtained from all tuberculosis patients prior to their enrollment in the study.
Results Demographic and clinical featuresOf the 149 TB patients with MDR Mycobacterium tuberculosis isolates, 109 (73.15%) were male, and 40 (26.85%) were female. The age of patients ranged from 14 to 88 years, with a mean age of 46.53 years.
The associations between demographic characteristics (sex and age), clinical features (treatment history and sputum smear results), and the lineage of MDR-MTB isolates were analyzed and are summarized in Table 1.

Table 1. Demographic and clinical features of patients infected with L4 and L2 lineage isolates.
Drug-resistance mutation spectrumTable 2 summarizes the drug-resistance mutation spectrum of 149 MDR-MTB isolates, highlighting mutations in resistant strains, while Supplementary Table S1 provides comprehensive details on the drug-resistance mutations, lineage information, and DST results.
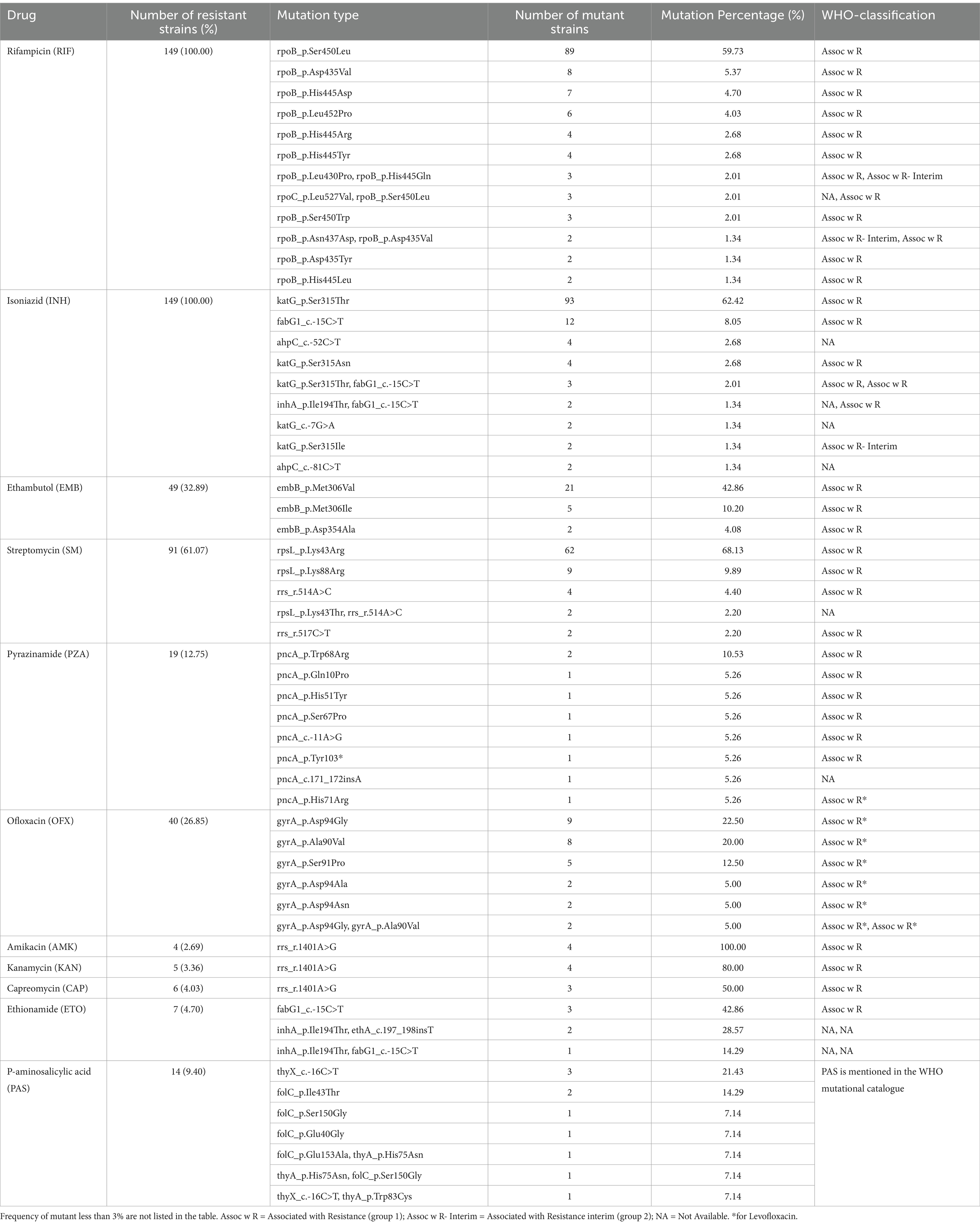
Table 2. Drug resistance gene mutation (for resistant isolates) spectrum of 149 MDR-MTB strains.
Rifampicin resistanceAmong 145 RIF-resistant isolates (97.32%, 145/149), 97.93% (142/145) harbored mutations in the rifampicin-resistance-determining region (RRDR) of the rpoB gene. The most prevalent mutation was rpoB Ser450Leu (59.73%), followed by Asp435Val, His445Asp., Leu452Pro, and others (Supplementary Table S1). Combined mutations at two sites were observed in 15 strains (10.07%, 15/149), while 5 strains (3.35%, 5/149) exhibited compensatory mutations in the rpoC gene (Supplementary Table S1).
Isoniazid (INH) resistanceMutations associated with INH resistance were detected in 138 strains (92.62%, 138/149), with 23 mutation types identified. The dominant mutation was katG Ser315Thr (62.42%, 93/149), followed by fabG1 c-15C > T, ahpC c-52C > T, and katG Ser315Asn. Mutations were mainly associated with katG (77.18%, 115/149) and fabG1 (15.44%, 23/149). Combined mutations were observed in 12 strains (8.05%, 12/149) (Supplementary Table S1).
Ethambutol resistanceAmong 49 EMB-resistant strains (26.53%, 49/149), 13 mutation types were identified. Common mutations included embB Met306Val (42.86%, 21/49) and Met306Ile (10.20%, 5/49). Combined mutations at two sites were observed in 3 strains (6.12%, 3/49) (Supplementary Table S1).
Streptomycin resistanceSM resistance was identified in 80 strains (87.91%, 80/91), with six mutation types primarily involving the rpsL and rrs genes. The most frequent mutations were rpsL Lys43Arg (68.13%, 62/91) and Lys88Arg (9.89%, 9/91). Combined mutations were observed in 2 strains (2.20%, 2/91) (Supplementary Table S1).
Pyrazinamide resistance resistancePZA resistance mutations were located in the pncA gene and its promoter region. Eight mutation types were identified, with pncA Trp68Arg (10.53%, 2/19) being the most frequent, followed by Gln10Pro, His51Tyr, Ser67Pro, and others.
Second-line drug resistanceSecond-Line Drug Resistance: Ofloxacin (OFX): Among 40 OFX-resistant strains, 10 mutation types were identified, with a high mutation rate in the quinolone-resistance-determining region (QRDR) of 96.88% (31/32).
Dominant mutations included gyrA Asp94Gly (22.50%, 9/40), Ala90Val (20.00%, 8/40), and Ser91Pro (12.50%, 5/40). Combined mutations were found in 4 strains (10.00%, 4/40).
Second-Line Injectable Drugs (AMK, KAN, CAP): Mutations primarily occurred in the rrs gene, with the 1401A>G mutation observed in all resistant strains: AMK (100.00%, 4/4), KAN (80.00%, 4/5), and CAP (50.00%, 3/6).
Ethionamide resistanceEthionamide (ETO): Mutations were identified in the fabG1, ethA, and inhA genes, with the most frequent being fabG1 -15C > T (42.86%, 3/7). Combined mutations were found in 3 strains (42.86%, 3/7).
Para-Aminosalicylic acid resistancePara-Aminosalicylic Acid (PAS): Mutations were detected in the thyA, folC, and thyX genes. The dominant mutation was thyX c-16C > T (21.43%, 3/14), with combined mutations observed in 3 strains (21.43%, 3/14).
Identification of the lineage and sublineageThis study identified two primary lineages among the 149 MDR-TB isolates. Lineage 4 (Euro-American genotype) accounted for 4.70% (7/149) of the isolates, while Lineage 2 (East Asian genotype) was predominant, comprising 95.30% (142/149). Among the isolates belonging to Lineage 2, the Beijing genotype (Lineage 2.2) was the dominant sublineage, representing 95.30% (142/149) of all isolates. In contrast, sublineages Lineage 4.2 and Lineage 4.4 were less common, accounting for 2.68% (4/149) and 2.01% (3/149), respectively. The Beijing genotype strains were further classified into two types: modern Beijing type, which constituted 88.03% (125/142) of the Beijing genotype isolates, and ancient Beijing type, which accounted for 11.97% (17/142). These findings highlight the predominance of the East Asian Lineage 2, particularly the modern Beijing genotype, among the MDR-TB isolates in this study.
Correlation between mutation in drug resistance-related genes and lineage/sublineageThe embB Met306Ile mutation was significantly more prevalent in lineage 4 compared to lineage 2 (p = 0.0150). In contrast, the rpsL Lys43Arg mutation occurred less frequently in lineage 4 (p = 0.0333) (Table 3).

Table 3. Distribution of main drug resistance gene mutation type in lineages and sublineages of 149 MDR-TB isolates in Wuhan.
Identification of the clustering featuresAnalysis of the SNP distance matrix revealed 14 clusters comprising a total of 31 MDR-MTB isolates (Figure 2), resulting in a clustering rate of 20.81% (31/149) and a minimum estimate of recent transmission of 11.41%. Each cluster included 2–3 MDR-MTB isolates. Among these clustered isolates, 21 originated from new MDR-TB patients, yielding a clustering rate of 22.11% (21/95), while 10 isolates came from previously treated MDR-TB patients, with a clustering rate of 18.52% (10/54).
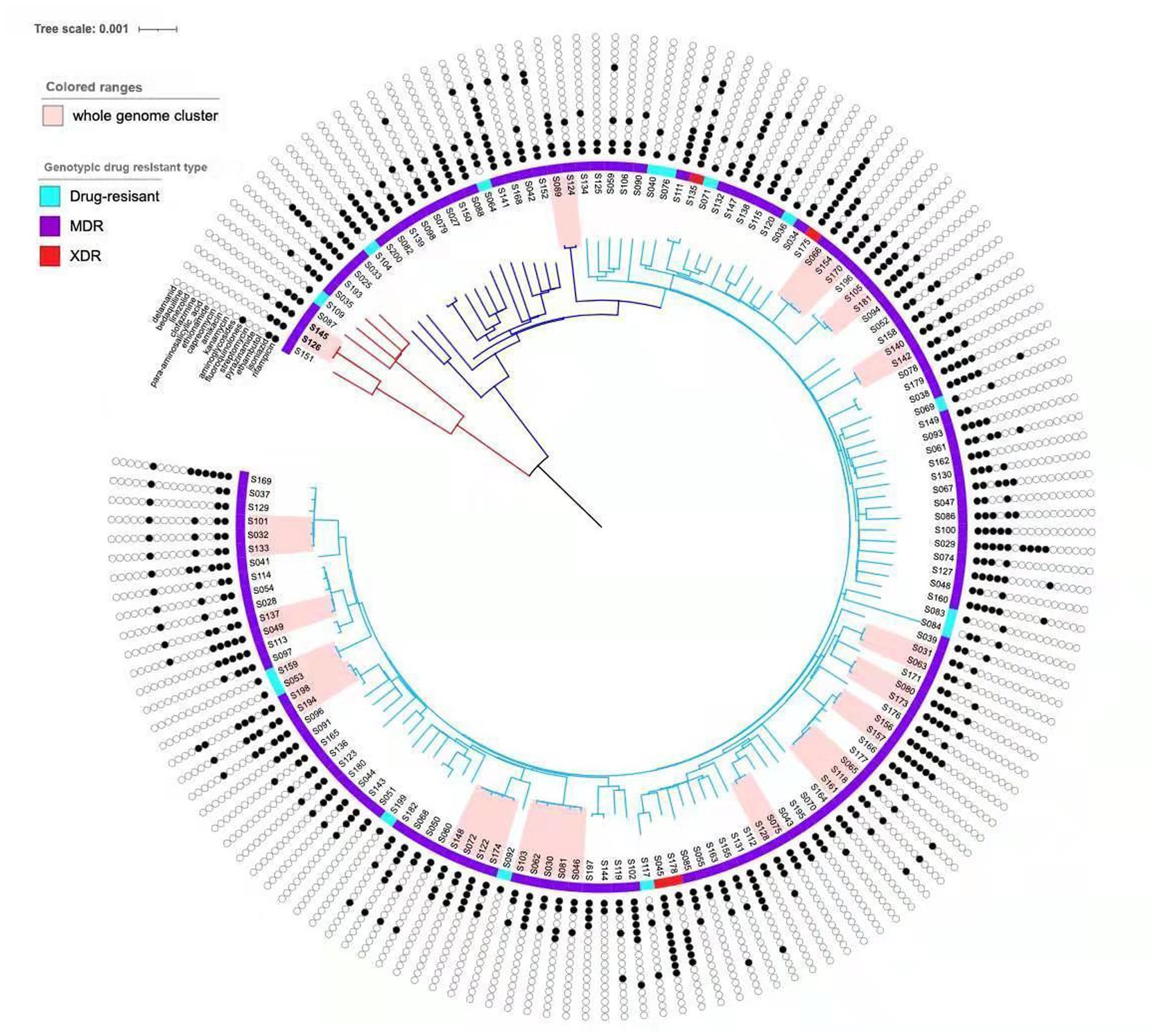
Figure 2. Phylogenetic tree of the 149 MTB isolates in this study. The sublineages, genotypic drug resistant type and drug resistant profile of the isolates are shown on the tree, according to the color legend shown on the left. The light pink bars indicate clustered isolates.
The clustering rate for lineage 4 (Euro-American) was 28.57% (2/7), compared to 20.42% (29/142) for lineage 2 (East Asian), though the difference was not statistically significant (χ2 = 0.0017, p = 0.9668) (Table 4). Within the clustered MDR-TB isolates, three pairs of patients resided within 2 kilometers of each other, including two pairs from the same household, suggesting direct transmission events.
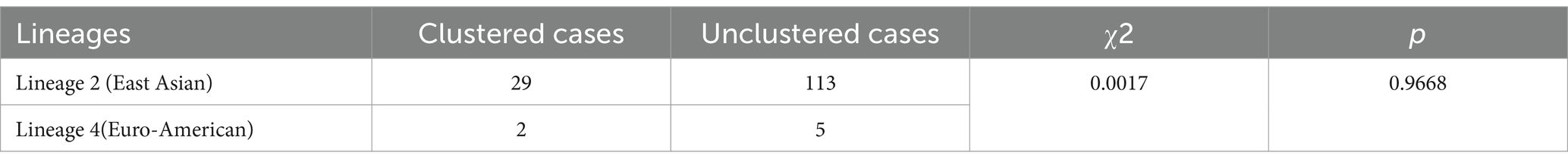
Table 4. Comparison of clustering rates between lineage 2 and lineage 4.
Consistency of genotypic and phenotypic resistance in MDR-MTB strainsWGS determines drug susceptibility by identifying mutations in resistance-related genes. If such mutations are detected, the strain is classified as genotypically resistant; otherwise, it is considered genotypically susceptible. However, the agreement between WGS-predicted resistance and phenotypic drug susceptibility testing is not absolute.
For RIF, the compliance rate between WGS and phenotypic testing was 97.32% (145/149). The compliance rate for INH was 92.62% (138/149), while for OFX, it was 87.92% (131/149). SM had a compliance rate of 83.89% (125/149), and EMB exhibited the lowest compliance rate at 71.14% (106/149) (Supplementary Table S1).
DiscussionIn recent years, WGS has proven to be a highly accurate method for predicting resistance to most first- and second-line anti-TB drugs, offering high sensitivity and specificity (18). Compared with targeted molecular resistance detection methods like GeneXpert MTB/RIF, WGS provides several advantages, including comprehensive detection of all drug resistance-associated mutations, identification of potential novel mutations, and detection of heterogeneous drug resistance.
Globally, the majority of studies have shown that mutations within the 81-bp rifampin-resistance-determining region (RRDR) of the rpoB gene account for over 96.00% of RIF resistance (19). In this study, 97.93% of RIF-resistance mutations were located within the RRDR. Mutations outside the RRDR are often undetected by molecular methods such as GeneXpert MTB/RIF. Notably, this study identified three mutations (rpoB Glu761Asp., Phe424Val, and Thr400Ala) outside the RRDR, which have not been previously reported in the literature. Additionally, five compensatory mutations related to RIF resistance were identified in the rpoC gene.
For INH resistance, mutations in the fabG1 and katG genes were detected in 92.62% (138/149) of MDR-MTB isolates, underscoring their pivotal role in conferring resistance. The most frequent mutation in katG was Ser315Thr (62.42%, 93/149), while the most common mutation in fabG1 was c-15C > T (8.05%, 12/149). A previously unreported katG mutation (−7G > A) was identified, along with a rare inhA Ile194Thr mutation, which had been documented in a prior Indian study (6). Furthermore, a novel ahpC mutation (c-52C > T) observed in this study was consistent with findings from southern Brazil (20). Nine compensatory mutations in the ahpC gene were also identified, highlighting their potential role in mediating INH resistance. These mutations have been reported in several other studies (20, 21).
For EMB resistance, the most prevalent mutations identified in this study were embB Met306Val (42.86%), Met306Ile, and Asp354Ala. The proportion of the embB Met306Val mutation observed here was lower than that reported in a Chinese study (22) but higher than the findings from an Ethiopian study (23). Additionally, a single mutation in the embA gene was detected in one strain; however, this mutation was not identified by the GenoType MTB DRplus Version 1 assay (Hain Lifescience, Germany).
Regarding SM resistance, the most frequent mutation among MDR-TB patients was rpsL Lys43Arg (68.13%), consistent with studies conducted in Taiwan (24), Hainan (25), China (22, 26), and southern China (27). Mutations rrs 514a > c and 517c > t were also reported in previous studies by Zhao (28) and Dorji (29). Furthermore, a novel mutation, rpsL Lys43Thr, identified in Mejía-Ponce’s study, was associated with reduced fitness (30).
Fluoroquinolones are essential in the treatment of MDR-TB and serve as one of the core drugs in treatment regimens. Resistance to quinolones primarily arises from mutations in the gyrA and gyrB genes (31). In this study, the most frequent mutation observed was gyrA Asp94Gly (22.50%, 9/40), consistent with findings from several studies conducted in China (32, 33).
For injectable aminoglycosides, the rrs 1401A > G mutation is a critical determinant of resistance. This study revealed mutation rates of 100.00, 80.00, and 50.00% for AMK, KAN, and CAP, respectively.
ETO resistance is well-documented to be associated with the fabG1 c-15C > T mutation (34). In this study, fabG1 c-15C > T and inhA mutations were the most prevalent, while one isolate exhibited an ethA mutation. The inhA Ile194Thr mutation, however, was not found in the WHO mutational catalog (35).
While PAS has been utilized in TB treatment for over 70 years, its mechanism of action remains incompletely understood (36). Resistance to PAS is generally linked to mutations in genes such as thyA, folC, and ribD (37). In this study, the most common mutation observed was thyX c-16C>T, corroborating findings from Srilohasin’s research (38). Additionally, folC Ser150Gly and Glu40Gly mutations were detected, similar to findings reported in Australia (29).
This study evaluated the efficacy of WGS in predicting drug resistance, using phenotypic DST as the reference standard. WGS demonstrated a high concordance rate (over 80.0%) for predicting resistance to RIF, INH, SM, and OFX, aligning with previous studies (39, 40). The discrepancy between WGS and phenotypic DST results may arise from several factors. First, phenotypic DST might fail to detect resistance in samples with low resistance levels (41). Second, WGS cannot capture resistance mechanisms beyond genetic mutations, such as those induced by efflux pumps (42). However, the accuracy of WGS in predicting EMB resistance was notably lower, consistent with other reports (25, 43). This lower accuracy may be due to single mutations, such as embA or embB M306I/G406A, being present in both EMB-resistant and EMB-sensitive strains (43, 44).
The genetic variations responsible for Mycobacterium tuberculosis drug resistance differ significantly across regions, influenced by factors such as national TB control policies, treatment strategies, population-related medical behaviors, and strain phylogenetic backgrounds (45–47).
In our study, the L2 and L4 lineages were the predominant strains among MDR-TB isolates from Wuhan, with L2 accounting for 95.30%. Of the L2 strains, 88.03% were classified as modern Beijing strains. The frequency of the embB Met306Ile mutation in L4 strains was higher than in L2 (p = 0.0150), while the frequency of the rpsL Lys43Arg mutation was lower in L4 strains compared to L2 (p = 0.0333). No significant differences were observed in mutation frequencies at other sites. The folC Ile43Thr mutation, associated with PAS resistance, was exclusively found in L2 strains, consistent with findings in other studies (26, 48, 49).
The Beijing genotype strains were further categorized into modern and ancient Beijing types (50, 51). Modern Beijing strains of MTB are more prevalent than ancient Beijing strains worldwide and have been responsible for the majority of TB outbreaks (52, 53). In our study, there were no significant differences in mutation types between modern and ancient Beijing strains, which contrasts with several previous studies (54), possibly due to the relatively small sample size. Additionally, the clustering rate between lineage 4 and lineage 2 strains did not show significant differences, consistent with studies conducted in Hainan (25) and Hunan (55), China.
WGS data of MDR-TB strains from Wuhan were used in this study to assess clustering based on SNP distance. A clustering rate of 20.81% was observed, with a minimum estimated proportion of recent transmission at 11.41%, which was lower than the findings reported by Yang (56) in Shanghai and similar to the results from Jiang (57) in Shenzhen.
The findings regarding Beijing family classification and drug-resistance-associated gene sequencing were consistent with our previous research (13). However, the clustering results differed: 38 cases were identified in 11 clusters using the DTM-PCR and MIRU-VNTR methods (13), whereas 31 cases were grouped into 14 clusters with WGS. Only one cluster (two cases) was identical across both genotyping methods. In this study, two groups of family members (each consisting of two individuals) were identified based on their residential addresses in the Wuhan TB reporting network, and both groups were clustered by WGS. In contrast, only one group was clustered by DTM-PCR and MIRU-VNTR (13).
Thus, the standard 24-locus MIRU-VNTR method has limitations due to the homology within strains of the Beijing family, and the combination of DTM-PCR and MIRU-VNTR did not adequately discriminate between strains to comprehensively trace transmission (12, 58, 59) provides more accurate gene clustering results in regions and countries where the Beijing family of Mycobacterium tuberculosis is predominant.
The recent transmission rate observed in this study may have been underestimated for several reasons. First, the MDR-TB patients enrolled were from Wuhan Pulmonary Hospital, excluding those from another designated hospital, Jin-yin-tan Hospital, which represents only approximately 40% of the total patient population in Wuhan. Second, the TB culture positivity rate in Wuhan in 2017 was only 40%, which may have limited the effectiveness of genotyping studies. Third, the study sample was collected in 2017, slightly earlier than anticipated due to delays caused by the Covid-19 pandemic. Consequently, newer anti-tuberculosis drugs, such as Bedaquiline and Delamanid, were not included in the treatment regimen. Furthermore, the epidemiological links among clustered patients were not investigated, partly due to loss of follow-up. As noted in previous studies (60, 61), the use of a single reference genome, H37Rv, may not adequately detect virulence-related loci in Mycobacterium tuberculosis. These five factors represent the main limitations of this study.
ConclusionThis study is the first to employ WGS to detect drug resistance-related gene mutations and assess the genetic clustering of MDR-MTB strains, thereby providing valuable insights into the molecular epidemiology of MDR-TB in Wuhan, China. The genetic diversity of MDR-TB in Wuhan mirrors global trends but also exhibits distinct regional characteristics. MDR-TB demonstrates localized transmission within Wuhan. WGS has proven to be an effective tool for investigating the molecular epidemiology of MDR-TB in this region. The findings of this study may serve as a critical reference for the prevention and clinical management of MDR-TB in Wuhan.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statementThe studies involving humans were approved by Wuhan Pulmonary Hospital Medical Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributionsLW: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing, Visualization. JC: Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Resources. ZD: Conceptualization, Data curation, Investigation, Methodology, Writing – review & editing. ZeZ: Conceptualization, Data curation, Investigation, Writing - review & editing. ZhZ: Formal analysis, Investigation, Methodology, Validation, Writing – original draft. QD: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Software, Writing – review & editing, Project administration, Supervision.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Project of Hubei Health Commission (grant no: WJ2021M026).
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2025.1442987/full#supplementary-material
Footnotes References3. Xie, C, Hu, X, Liu, Y, and Shu, C. Performance comparison of GeneXpert MTB/RIF, Gene Chip Technology, and modified Roche culture method in detecting mycobacterium tuberculosis and drug susceptibility in sputum. Contrast Media Mol Imaging. (2022) 2022:2995464. doi: 10.1155/2022/2995464
PubMed Abstract | Crossref Full Text | Google Scholar
4. Sánchez-Cabral, O, Santillán-Díaz, C, Flores-Bello, ÁP, Herrera-Ortega, MI, Sandoval-Gutiérrez, JL, Santillán-Doherty, P, et al. GeneXpert(®) MTB/RIF assay with transbronchial lung cryobiopsy for Mycobacterium tuberculosis diagnosis. Ann Transl Med. (2020) 8:351. doi: 10.21037/atm.2020.02.100
Crossref Full Text | Google Scholar
5. Vīksna, A, Sadovska, D, Berge, I, Bogdanova, I, Vaivode, A, Freimane, L, et al. Genotypic and phenotypic comparison of drug resistance profiles of clinical multidrug-resistant Mycobacterium tuberculosis isolates using whole genome sequencing in Latvia. BMC Infect Dis. (2023) 23:638. doi: 10.1186/s12879-023-08629-7
Crossref Full Text | Google Scholar
6. Tamilzhalagan, S, Shanmugam, S, Selvaraj, A, Suba, S, Suganthi, C, Moonan, PK, et al. Whole-genome sequencing to identify missed rifampicin and isoniazid resistance among tuberculosis isolates-Chennai, India, 2013-2016. Front Microbiol. (2021) 12:720436. doi: 10.3389/fmicb.2021.720436
Crossref Full Text | Google Scholar
7. Shibabaw, A, Gelaw, B, Ghanem, M, Legall, N, Schooley, AM, Soehnlen, MK, et al. Molecular epidemiology and transmission dynamics of multi-drug resistant tuberculosis strains using whole genome sequencing in the Amhara region, Ethiopia. BMC Genomics. (2023) 24:400. doi: 10.1186/s12864-023-09502-2
Crossref Full Text | Google Scholar
8. Han, Z, Li, J, Sun, G, Gu, K, Zhang, Y, Yao, H, et al. Transmission of multidrug-resistant tuberculosis in Shimen community in Shanghai, China: a molecular epidemiology study. BMC Infect Dis. (2021) 21:1118. doi: 10.1186/s12879-021-06725-0
Crossref Full Text | Google Scholar
9. Beggs, ML, Eisenach, KD, and Cave, MD. Mapping of IS6110 insertion sites in two epidemic strains of Mycobacterium tuberculosis. J Clin Microbiol. (2000) 38:2923–8. doi: 10.1128/jcm.38.8.2923-2928.2000
Crossref Full Text | Google Scholar
10. Kamerbeek, J, Schouls, L, Kolk, A, van Agterveld, M, van Soolingen, D, Kuijper, S, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. (1997) 35:907–14. doi: 10.1128/jcm.35.4.907-914.1997
Crossref Full Text | Google Scholar
11. Supply, P, Allix, C, Lesjean, S, Cardoso-Oelemann, M, Rüsch-Gerdes, S, Willery, E, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. (2006) 44:4498–510. doi: 10.1128/jcm.01392-06
Crossref Full Text | Google Scholar
12. Coll, P, and García De Viedma, D. Molecular epidemiology of tuberculosis. Enferm Infecc Microbiol Clin (Engl Ed). (2018) 36:233–40. doi: 10.1016/j.eimc.2018.01.001
Crossref Full Text | Google Scholar
13. Duan, Q, Zhang, Z, Tian, D, Zhou, M, Hu, Y, Wu, J, et al. Transmission of multidrug-resistant Mycobacterium tuberculosis in Wuhan, China: a retrospective molecular epidemiological study. Medicine (Baltimore). (2022) 101:e28751. doi: 10.1097/md.0000000000028751
Crossref Full Text | Google Scholar
15. Comín, J, Cebollada, A, Ibarz, D, Viñuelas, J, Sahagún, J, Torres, L, et al. Analysis of the twenty-six largest outbreaks of tuberculosis in Aragon using whole-genome sequencing for surveillance purposes. Sci Rep. (2022) 12:18766. doi: 10.1038/s41598-022-23343-1
PubMed Abstract | Crossref Full Text | Google Scholar
16. Sun, H, Ma, Z, Ai, F, Han, B, Li, P, Liu, J, et al. Insidious transmission of Mycobacterium tuberculosis in Ordos, China: a molecular epidemiology study (2024) 43:305–12. doi: 10.1007/s10096-023-04730-6,
Crossref Full Text | Google Scholar
17. Mestre, O, Luo, T, Dos Vultos, T, Kremer, K, Murray, A, Namouchi, A, et al. Phylogeny of Mycobacterium tuberculosis Beijing strains constructed from polymorphisms in genes involved in DNA replication, recombination and repair. PLoS One. (2011) 6:e16020. doi: 10.1371/journal.pone.0016020
Crossref Full Text | Google Scholar
18. Che, Y, Lin, Y, Yang, T, Chen, T, Sang, G, Chen, Q, et al. Evaluation of whole-genome sequence to predict drug resistance of nine anti-tuberculosis drugs and characterize resistance genes in clinical rifampicin-resistant Mycobacterium tuberculosis isolates from Ningbo, China. Front Public Health. (2022) 10:956171. doi: 10.3389/fpubh.2022.956171
PubMed Abstract | Crossref Full Text | Google Scholar
19. Jaksuwan, R, Tharavichikul, P, Patumanond, J, Chuchottaworn, C, Chanwong, S, Smithtikarn, S, et al. Genotypic distribution of multidrug-resistant and extensively drug-resistant tuberculosis in northern Thailand. Infect Drug Resist. (2017) 10:167–74. doi: 10.2147/idr.s130203
Crossref Full Text | Google Scholar
20. Salvato, RS, Reis, AJ, Schiefelbein, SH, Gómez, MAA, Salvato, SS, da Silva, LV, et al. Genomic-based surveillance reveals high ongoing transmission of multi-drug-resistant Mycobacterium tuberculosis in southern Brazil. Int J Antimicrob Agents. (2021) 58:106401. doi: 10.1016/j.ijantimicag.2021.106401
PubMed Abstract | Crossref Full Text | Google Scholar
21. Napier, G, Campino, S, Phelan, JE, and Clark, TG. Large-scale genomic analysis of Mycobacterium tuberculosis reveals extent of target and compensatory mutations linked to multi-drug resistant tuberculosis. Sci Rep. (2023) 13:623. doi: 10.1038/s41598-023-27516-4
PubMed Abstract | Crossref Full Text | Google Scholar
22. Wan, L, Liu, H, Li, M, Jiang, Y, Zhao, X, Liu, Z, et al. Genomic analysis identifies mutations concerning drug-resistance and Beijing genotype in multidrug-resistant Mycobacterium tuberculosis isolated from China. Front Microbiol. (2020) 11:1444. doi: 10.3389/fmicb.2020.01444
Crossref Full Text | Google Scholar
23. Welekidan, LN, Yimer, SA, Skjerve, E, Dejene, TA, Homberset, H, Tonjum, T, et al. Whole genome sequencing of drug resistant and drug susceptible Mycobacterium tuberculosis isolates from Tigray region, Ethiopia. Front Microbiol. (2021) 12:743198. doi: 10.3389/fmicb.2021.743198
PubMed Abstract | Crossref Full Text | Google Scholar
24. Xiao, YX, Liu, KH, Lin, WH, Chan, TH, and Jou, R. Whole-genome sequencing-based analyses of drug-resistant Mycobacterium tuberculosis from Taiwan. Sci Rep. (2023) 13:2540. doi: 10.1038/s41598-023-29652-3
Crossref Full Text | Google Scholar
25. Wang, J, Yu, C, Xu, Y, Chen, Z, Qiu, W, Chen, S, et al. Analysis of drug-resistance characteristics and genetic diversity of multidrug-resistant tuberculosis based on whole-genome sequencing on the Hainan Island, China. Infect Drug Resist. (2023) 16:5783–98. doi: 10.2147/idr.s423955
Crossref Full Text | Google Scholar
26. Gao, M, Yang, TT, Li, GL, Chen, R, Liu, HC, Gao, Q, et al. Analysis on drug resistance-associated mutations of multi-drug resistant Mycobacterium tuberculosis based on whole-genome sequencing in China. Zhonghua Liu Xing Bing Xue Za Zhi. (2020) 41:770–5. doi: 10.3760/cma.j.cn112338-20191111-00800
PubMed Abstract | Crossref Full Text | Google Scholar
27. Islam, MM, Tan, Y, Hameed, HMA, Chhotaray, C, Liu, Z, Liu, Y, et al. Phenotypic and genotypic characterization of streptomycin-resistant multidrug-resistant Mycobacterium tuberculosis clinical isolates in southern China. Microb Drug Resist. (2020) 26:766–75. doi: 10.1089/mdr.2019.0245
Crossref Full Text | Google Scholar
28. Zhao, LL, Huang, MX, Xiao, TY, Liu, HC, Li, MC, Zhao, XQ, et al. Prevalence, risk and genetic characteristics of drug-resistant tuberculosis in a tertiary care tuberculosis hospital in China. Infect Drug Resist. (2019) 12:2457–65. doi: 10.2147/idr.s209971
留言 (0)