Cell culture and Eltrombopag treatment
HuR knockout (KO) HeLa cell lines were previously generated using CRISPR-Cas9 as described [22]. Wild-type and HuR KO HeLa cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) + GlutaMAX with 10% fetal bovine serum (FBS) (Gibco). Recombinant HuR with mCherry protein was expressed as reported previously [22]. Eltrombopag was purchased from Selleckchem (catalogue number S4502). The viability of HeLa wild-type (WT) and HuR knockout (KO) cells treated with 20 µM of Eltrombopag or DMSO was assessed using CellTiterGlo at various time points (0 h, 3 h, 6 h, 12 h, 24 h, and 48 h) using CellTiter-Glo 3D Viability Assay, following the manufacturer’s protocol.
RNA pull-down confocal nanoscanning (RP-CONA)
The interaction between HuR and DHX9 protein and Eltrombopag was evaluated by RNA pull-down confocal nanoscanning (RP-CONA) assay as described before [22]. Briefly, a 5′ 6-FAM- and 3′ biotin-labeled RNA pri-miR-7 conserved terminal loop (CTL) transcript (pri-miR-7–6-FAM) (IDT) was immobilized on streptavidin-coated Ni–NTA agarose beads (ProteoGenix). Extracts from HuR KO HeLa cells expressing mCherry-HuR were incubated with RNA-bound beads and various concentrations of Eltrombopag (0, 10, 20, 30, 40, and 50 μM). After incubation, beads were imaged by confocal microscopy (Opera Phoenix) to quantify mCherry-HuR recruitment to fluorescently labeled RNA. The RP-CONA between mCherry-DHX9 from HeLa extracts and pri-miR-7–6-FAM with DMSO or Eltrombopag (20 μM) was used as a negative control. Data were obtained from three technical replicates and mean was calculated. For fluorescence anisotropy, the IC50 was calculated using nonlinear regression (4PL, GraphPad Prism Software 10.0.2, Radj2 = 0.98).
RNA extraction and quality control
Cells were seeded in 6-well plates and treated with DMSO (control) and Eltrombopag (20 μM). After 48 h, total RNA was extracted from two replicates per treatment using TRIzol reagent (Life Technologies, CA, USA). RNA concentration was quantified using Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and RNA integrity was checked using Agilent 5200 Fragment Analyzer (Agilent Technologies, Palo Alto, CA, USA). The samples with RNA integrity number (RIN) ≥ 9 were subjected to the subsequent analysis. Initially we prepared two set of samples for RNAseq analysis (batch 1) followed by an additional sample set (batch 2).
RNA sequencing and data analysis
A total of 2 μg of total RNA per sample was used as input material for the RNA library. Sequencing libraries were generated using standard library for IlluminaR (NEB, USA) following the manufacturer’s recommendations.
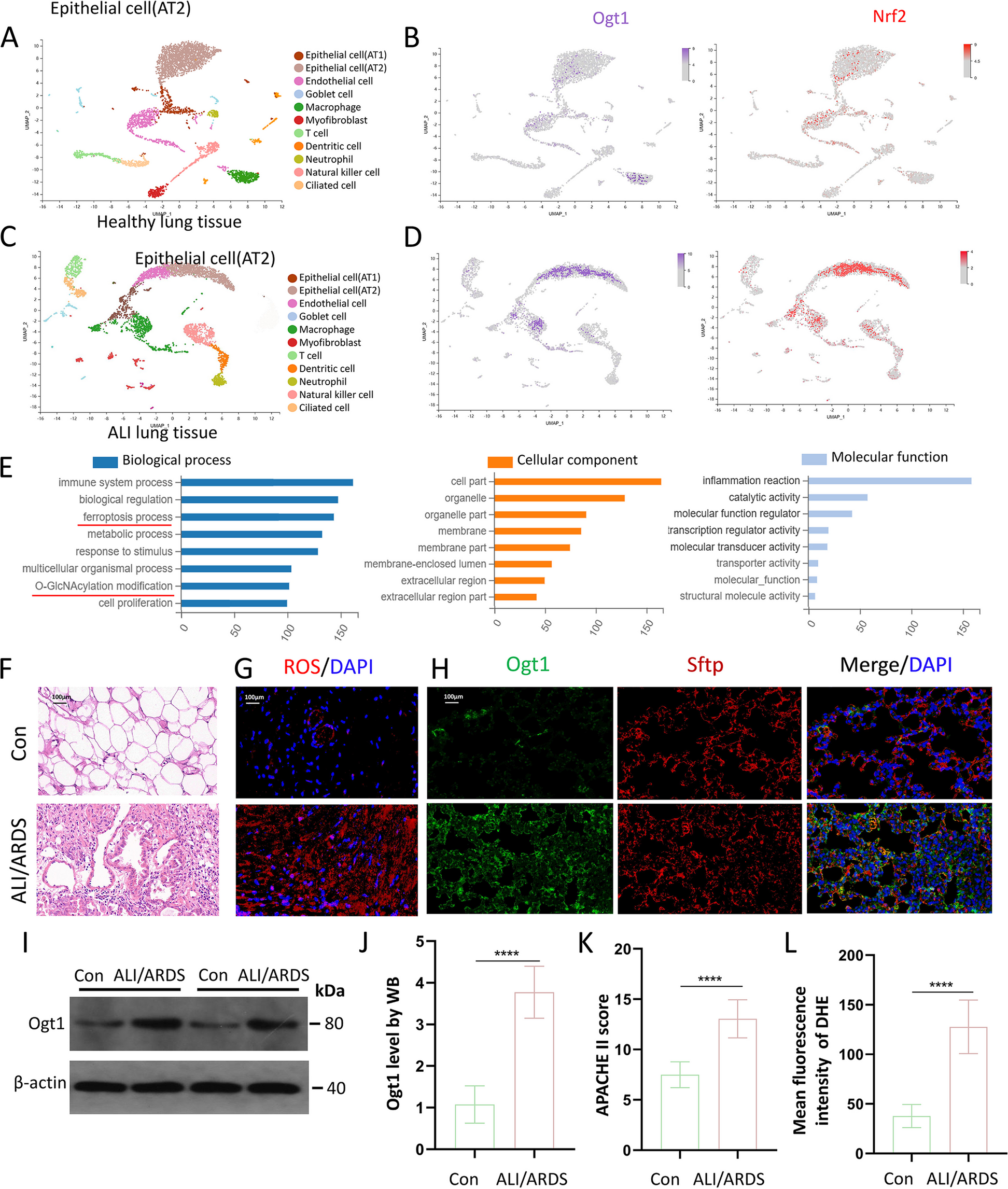
Samples were sequenced on Illumina NovaSeq 6000 to generate 150 nucleotide reads with paired end. Raw sequence data was converted into fastq files and de-multiplexed using bcl2fastq 2.20, allowing for one mismatch in index sequence identification. Quality control of raw reads was performed using fastqc, and reads were trimmed using BBDuk. Cleaned paired-end reads were then mapped to the human reference genome using the STAR aligner v.2.5.2b [56]. Gene quantification was performed on the resulting BAM files using Salmon with the help of genome index build based on GENCODE release 42 to obtain unique gene counts. Differential gene expression analysis was performed between treatment groups using DESeq2 [57]. Genes with log2FC ± 1 and adjusted p values < 0.05 were considered as statistically significant for each comparison. Gene-set enrichment analysis on differently expressed genes were performed using ShinyGO 0.80 [58]; the data visualization was done using ggplot2 package. Volcano plots, principal component analysis (PCA), sample-to-sample distances, and Euler diagrams were performed to demonstrate changes and patterns within each comparison among all samples using in-house R and Python scripts. AREs were downloaded from the ARED-plus and compared with ENSEMBL ID of DE genes [59].
Mass spectrometry
Cells were seeded in 6-well plates and incubated overnight to adhere. The next day, they were treated with DMSO (control) and Eltrombopag (20 µM). After 48 h, protein from 3 replicates have been collected. Protein was extracted using Roeder D (no glycerol), regular sonication cycle. Protein samples from all biological replicates were processed at the same time and with using the same digestion protocol without any deviations. They were subjected for MS analysis under the same conditions. Protein and peptide lists generated using the same software and the same parameters. Specifically, 5 µg of total protein from each sample were digested using the Filter Aided Sample Preparation (FASP) protocol as described by Wisniewski et al. with minor modifications [60]. In brief, each protein sample was added on the top of a 30 kDa MWCO filter units (Vivacon, UK) along with 150 µl of denaturation buffer (8 M urea in 50 mM ammonium bicarbonate (ABC) (Sigma Aldrich)) and spun at 14,000 × g for 20 min, while another wash with 200 µl of denaturation buffer was performed under the same conditions. The protein samples were then reduced by the addition of 100 µl of 10 mM dithiothreitol (Sigma Aldrich, UK) in denaturation buffer for 30 min at ambient temperature, and alkylated by adding 100 µl of 55 mM iodoacetamide (Sigma Aldrich, UK) in denaturation buffer for 20 min at ambient temperature in the dark. Two washes with 100 µl of denaturation buffer and two with digestion buffer (50 mM ABC) were performed under the same conditions described above before the addition of trypsin (Pierce, UK). The protease:protein ratio was 1:50 and proteins were digested overnight at 37 °C. Following digestion, samples were spun at 14,000 × g for 20 min and the flow-through containing digested peptides was collected. Filters were then washed one more time with 100 µl of digestion buffer and the flow-through was collected again. The eluates from the filter units were acidified using 20 µl of 10% trifluoroacetic acid (TFA) (Sigma Aldrich) and spun onto StageTips as described before [61]. Peptides were eluted in 40 μl of 80% acetonitrile in 0.1% TFA and concentrated down to 1 μl by vacuum centrifugation (Concentrator 5301, Eppendorf, UK). The peptide sample was then prepared for LC–MS/MS analysis by diluting it to 5 μl by 0.1% TFA.
LC–MS analyses were performed on an Orbitrap Exploris™ 480 Mass Spectrometer (Thermo Fisher Scientific, UK) coupled on-line to an Ultimate 3000 HPLC (Dionex, Thermo Fisher Scientific, UK). Peptides were separated on a 50 cm (2 µm particle size) EASY-Spray column (Thermo Scientific, UK), which was assembled on an EASY-Spray source (Thermo Scientific, UK) and operated constantly at 50 °C. Mobile phase A consisted of 0.1% formic acid in LC–MS grade water and mobile phase B consisted of 80% acetonitrile and 0.1% formic acid. Peptides were loaded onto the column at a flow rate of 0.3 μl min−1 and eluted at a flow rate of 0.25 μl min−1 according to the following gradient: 2 to 40% mobile phase B in 180 min and then to 95% in 11 min. Mobile phase B was retained at 95% for 5 min and returned back to 2% a minute after until the end of the run (220 min).
Survey scans were recorded at 120,000 resolution (scan range 350–1650 m/z) with an ion target of 5.0e6 and injection time of 20 ms. MS2 Data Independent Acquisition (DIA) was performed in the Orbitrap at 30,000 resolutions with a scan range of 200–2000 m/z, maximum injection time of 55 ms, and AGC target of 3.0E6 ions. We used HCD fragmentation [62] with stepped collision energy of 25.5, 27, and 30. The inclusion mass list with the correspondent isolation windows are shown in the table below. Data for both survey and MS/MS scans were acquired in profile mode.
MS data were searched against the UniProt human database using MaxQuant v1.6.6 [63]. Label-free quantification was performed using the MaxLFQ algorithm [64] integrated into MaxQuant. Differential gene expression analysis was performed using DEqMS with cut offs (log2FC ± 1 and padj < 0.05) [65]. Insignificant proteins of HuR KO treatment were correlated and cross compared with knockout effect and WT treatment effect.
qRT-PCR
Total RNA was extracted from HeLa cells (wild type, WT-E20, HuR KO, HuR KO E20) using the InviGen Invisorb spin virus RNA mini kit (Invitek) following the manufacturer’s protocol. RNA concentration and purity were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific). Total RNA was reverse transcribed into cDNA using the Promega 1 Step qRT-PCR kit (Promega) per the manufacturer’s instructions. Twenty-five-microliter reactions were prepared and qRT-PCR was performed using the QuantStudio 5 Real-Time PCR System (Applied Biosystems). Three biological replicates and three technical replicates were analyzed for each sample. QuantStudio Design and Analysis Software v1.5.2 (Applied Biosystems) was used to determine Cq values. Relative quantification of gene expression was performed using the comparative CT method (ΔΔCT method) in R programming language. GAPDH and ACTNB were used as reference genes for normalization. Throughout our study, statistical significance was determined by Student t-test comparing WT and treatment groups using R. p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). Data are denoted as mean ± SEM. The data are presented as mean ± S.D., with n ≥ 3 per group per treatment for all studies. One-way ANOVA was implemented to find statistical significance between scramble Eltrombopag treated against HuR KD Eltrombopag treated cells. The primer sequences are listed in Additional file 1: Table S2.
miR-7 measurement
HeLa WT cells were treated with DMSO or 20 µM Eltrombopag, and total RNA was collected at multiple time points (0, 3, 6, 12, 24, and 48 h) using the Total RNA Zol-Out D kit. RNA was reverse transcribed into cDNA using the microScript microRNA cDNA Synthesis Kit (Cat# 54,410). Equal amounts of cDNA were subjected to qPCR, using the Promega 1-Step qRT-PCR kit (Promega) per the manufacturer’s instructions. Three biological replicates and three technical replicates were analyzed for each sample. QuantStudio Design and Analysis Software v1.5.2 (Applied Biosystems) was used to determine Cq values. Relative quantification of gene expression was performed using the comparative CT method (ΔΔCT method).
Western blot analysis
Cultured cells were washed once with PBS and lysed in RIPA buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 7.4, 150 mM NaCl, 0.5 mM EDTA) supplemented with 100 × Protease Inhibitor Cocktail (P8340; Sigma-Aldrich) and Phosphatase Inhibitor Cocktail 2 (P5726; Sigma-Aldrich) and 3 (P0044; Sigma-Aldrich). After centrifugation (13,000 rpm, 15 min, 4 °C), the supernatants were collected. Protein concentration was determined with BCA Protein Assay Kit (23,225; Thermo Fisher Scientific). Thirty micrograms of total protein per sample was mixed with 6 μl of 5 × SB buffer (250 mM Tris–HCl, 10% SDS, 50% glycerol, 250 mM DTT, 0.02% bromophenol blue), loaded on 12% SDS PAGE and transferred onto a nitrocellulose membrane. Membranes were blocked for 1 h with Western Blocking Reagent (Roche), 5% milk or 5% BSA (Sigma) in TBST and incubated overnight at 4 °C with specific primary antibodies (Additional file 1: Table S3). After 1-h incubation with horseradish peroxidase-conjugated secondary antibody (P044801-2; Agilent), the signal was detected with Clarity Western ECL Substrate (179–5061; Bio-Rad) and visualized with ChemiDoc imaging system (Bio-Rad). Densitometric analysis was performed using Image Lab software (Bio-Rad).
Fluorescence anisotropy
Fluoresce anisotropy experiments were carried out in a buffer containing phosphate buffer pH = 7.5, 25 mM MgCl, 250 mM NaCl, 0.1% Tween 20, and 5 mM DTT. The concentration of pri-miR-7–1–6-FAM CTL (IDT) was 20 nM, Recombinant Human Elav-like protein 1 (ELAVL1/HuR) (CusaBio) was 500 nM, and concentrations of Eltrombopag were 0, 0.625, 1.25, 2.5, 5, 10, 20, 40, 60, 80, and 100 µM. Samples were prepared in triplicates in a black 384-well assay plate with round bottom. Fluorescence anisotropy was measured using Tecan INFINITE M1000 operated by Magellan software. Excitation and emission wavelengths were 495 nm and 520 nm. IC50 of Eltrombopag was calculated with a non-linear regression model using GraphPad Prism Software 10.0.2.
RNAi
HeLa cells were seeded at a density of 300,000 cells per well in 6-well plates, allowing them to reach about 60% confluency. The cells were left overnight to attach under standard culture conditions. The next day, they were gently washed once with fresh growth media and then with Opti-MEM (Thermo Fisher Scientific). Afterward, 1.5 ml of DMEM was added to each well. For transfection, a mixture of lipofectamine and siRNA (anti-HuR or scramble control) was prepared according to the manufacturer’s protocol (RNAiMAX, Invitrogen) and added to the cells in a total volume of 0.5 ml, with a final siRNA concentration of 100 pmol per well. The siRNA was sourced from Horizon Discovery. After 24 h of transfection, the media was removed, and the cells were washed with fresh media. The cells were then treated with 20 µM Eltrombopag (Selleckchem), while DMSO served as the negative control. After 48 h of treatment, the cells were collected for analysis. RNA samples were extracted using TRIzol reagent (Thermo Fisher Scientific) from wells dedicated to qRT-PCR, while protein samples were prepared by lysing cells with RIPA buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific) for western blot analysis.
Polysome profiling
Polysome fractionation was performed as described in the protocol [66]. Briefly, 48 h prior to fractionation, cells were treated with either DMSO (control) or 20 μM Eltrombopag. The day before cell lysis, 10–50% sucrose gradients were prepared in GB buffer (10 mM HEPES–KOH pH 7.2, 150 mM KCl, 5 mM MgCl2, Protease Inhibitor Cocktail (Roche), 100 µg/ml cycloheximide—CHX (J66901.03, Thermo Fisher Scientific), 4 U/ml RiboLock (EO0382, Thermo Fisher Scientific), and nuclease-free water). On the day of lysis, cells were treated with 100 µg/ml CHX, and 15 × 10^6 cells were lysed in GB buffer supplemented with 1% NP40 for 10 min on ice. After centrifugation at 14,000 rpm for 5 min at 4 °C, the protein concentration in the supernatants was measured using a NanoDrop spectrophotometer. Equal amounts of proteins were loaded onto the top of the sucrose gradients, and the gradients were subjected to ultracentrifugation at 38,000 rpm for 2 h at 4 °C using an Optima XPN ultracentrifuge (RRID: SCR_018238) and an SW41Ti rotor (Beckman Coulter). Following centrifugation, sucrose fractions were collected using a Density Gradient Fractionation System (Teledyne ISCO) with a Foxy Jr. Fraction Collector. Fractions were stored at − 80 °C for subsequent analysis. During collection, absorbance at 254 nm was monitored and recorded to generate RNA distribution profiles, which were printed on ISCO paper, scanned, and digitized using PlotDigitizer (https://plotdigitizer.com). Graph generation was performed using GraphPad Prism 10.
For HuR protein distribution in the sucrose fractions, 26 µl of each fraction was mixed with 4 µl of 5 × SB buffer, boiled, and subjected to SDS-PAGE. Western blot analysis was then performed, and signal intensities were quantified via densitometry. The total signal intensity across all fractions was normalized to 100%, and the percentage of signal intensity for each fraction was calculated.
For mRNA distribution analysis, 50 µl from each fraction was pooled as depicted in Additional file 1: Fig. S10A, mixed with 800 µl of TRIzol reagent and supplemented with 2 µl of TATAA Universal RNA Spike II (RS25SII, TATAA Biocenter). RNA was extracted according to the manufacturer’s protocol and purified using columns (A&A Biotechnology, 031–100). RNA was eluted with equal volumes of water from each column, and RNA purity was assessed using a NanoDrop spectrophotometer. qRT-PCR analysis was performed to quantify mRNA abundance in each fraction, and the percent distribution of mRNA across the gradients was calculated using the ΔΔCT method, with normalization to TATAA Universal RNA Spike values.
HuR ribonucleoprotein immunoprecipitation (RIP)
RIP analysis of HuR protein was performed according to the protocol described [67]. Briefly, 25 µl of Protein G Dynabeads (Invitrogen, 10003D) was incubated with 2.5 µg of mouse monoclonal anti-HuR antibody (Santa Cruz Biotechnology, sc-5261) or normal mouse IgG1 isotype antibody (Santa Cruz Biotechnology, sc-3877) for 1 h at room temperature (RT) in NT2 buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40). After 48 h of treatment with DMSO (control) or 20 µM Eltrombopag (E20), cells were lysed in PEB buffer (20 mM Tris–HCl, pH 7.5, 100 mM KCl, 5 mM MgCl2, 0.5% NP-40) supplemented with a Protease Inhibitor Cocktail and 40 U/ml of RiboLock. A total of 1000 µg of protein lysate was added to the antibody-coated bead complexes, and the immunoprecipitation (IP) reaction was carried out in NT2 buffer (supplemented with 10 mM DTT, 400 U/ml RiboLock, 15 mM EDTA, and 0.2% DMSO or 20 µM Eltrombopag) for 2 h at 4 °C and 30 min at RT with rotation. The RIP complexes were washed five times with 1 ml of ice-cold NT2 buffer containing 40 U/ml RiboLock. A 100 µl aliquot of the RIP-bead complexes from the final wash was taken for western blot analysis to verify the efficiency of the immunoprecipitation. Following the final wash, the RIP complexes were treated with DNase I (10 U/µl) in 100 µl of NT2 buffer for 10 min at 37 °C to degrade any genomic DNA. The complexes were then washed once more in NT2 buffer. To release HuR-bound RNAs, proteinase K treatment was performed in NT2 buffer supplemented with 0.5 µg/µl proteinase K (Thermo Fisher Scientific, EO0491) and 0.1% SDS. The supernatants containing the released RNAs were collected and mixed with TRIzol reagent for RNA extraction, following the manufacturer’s protocol. RNA was then purified using spin columns. RNA purity was assessed using a NanoDrop spectrophotometer. qRT-PCR was performed to quantify mRNA enrichment in the RIP samples. The results were calculated using the ΔΔCT method, with normalization to GAPDH mRNA.
Proteasome inhibition
HeLa WT cells were treated with Mg132 [5 µM] (474,790 Sigma) and (or) Eltrombopag [20 µM] for 24 h. The protein levels were determined with western blot technique as previously described. Densitometric analysis was performed using Image Lab software (Bio-Rad).
Functional assays and flow cytometry
Intracellular reactive oxygen species (ROS) levels were measured using CellROX Deep Red reagent (Invitrogen, C10422) according to manufacturer’s protocol. Cells were analyzed by flow cytometry on a CytoFLEX instrument (Beckman Coulter) using the APC channel. Intracellular ferrous iron (Fe2 +) was measured by FerroOrange staining (Dojindo, F374) and flow cytometry using the PE channel. Lipid peroxidation was assessed with the Lipid Peroxidation Assay Kit (Abcam, ab243377) per kit instructions. Oxidized lipid levels were detected by flow cytometry by comparing FITC and PE channel. In all experiments, cell viability was determined by staining with LIVE/DEAD Fixable Aqua or Violet dyes (Invitrogen, L34966/L34964). Data was acquired on CytoFLEX and analyzed using CytExpert software (Beckman Coulter). Geometric mean fluorescence intensity (MFI) was quantified for each probe. In our experiments, we adopted a comparative approach, using wild-type (WT) cells as the baseline control for the study. Specifically, WT cells were analyzed as controls for HuR KO cells to evaluate the effects of HuR deletion on the specified cellular responses. Additionally, HuR KO cells treated with Eltrombopag served as controls for WT cells exposed to Eltrombopag, enabling us to distinguish between HuR-dependent and HuR-independent effects of the treatment. Statistical significance for the HuR KO and Eltrombopag treatment factors was determined using a two-way ANOVA. For fluorescent probes, we built upon previously established and published methods, which we had successfully optimized and applied for flow cytometric measurements of ROS, lipid peroxides, and labile iron using the respective probes [68].









留言 (0)