
記住我





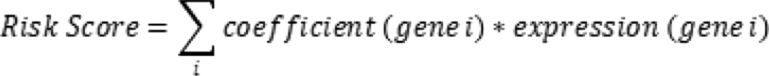


To investigate the landscape of solute carrier (SLC) family genes in osteosarcoma, we analyzed single-cell RNA sequencing (scRNA-seq) datasets from GSE152048 and GSE162454. After quality control, a total of 110,364 cells were retained for further examination. Dimensionality reduction and clustering revealed eight major cell lineages: B cells, endothelial cells, fibroblasts, myeloid cells, NK/T cells, osteoclasts, osteosarcoma (OS) cells, and pericytes (Fig. 1A). The proportion of these cell types varied across samples (Fig. 1B). Bubble plots displayed scaled expression levels and the proportion of specific markers for each cell type (Fig. 1C), while volcano plots illustrated significantly differentially expressed genes among the lineages (Fig. 1D). Utilizing CellChat analysis, we identified diverse interactions between different cell types (Fig. 1E, F).
Fig. 1
Single-cell landscape in osteosarcoma. (A) t-SNE plot of single cells profiled in osteosarcoma. (B) Cell proportions in each samples. (C) Bubble plot of the average and percent expression of different markers in each cell types. (D) Differential gene expression analysis reveals up and down-regulated genes across various cell types. An adjusted p value < 0.01 is indicated in red, while an adjusted p value ≥ 0.01 is indicated in black. (E) Network of cell-cell communication; Number of interactions (top); Strength of interactions (bottom). (F) Heatmap of cell-cell communication; Number of interactions (left); Strength of interactions (right)
The single-cell sequencing data revealed distinct expression patterns of solute carrier (SLC) superfamily genes across different cell types (Fig. 2A, B). Using the AddModuleScore algorithm, we calculated SLC scores for each cell type, incorporating all 449 SLC genes as a gene set. Unexpectedly, osteoclasts had the highest SLC scores, followed by myeloid cells and B cells, then OS cells, while other cell types exhibited lower scores (Fig. 2C). Based on the metabolic pathways of the substrates they transport, we categorized the SLCs into four groups: Carbohydrate Metabolism SLC (CM-SLC), Lipid Metabolism SLC (LM-SLC), Amino Acid Metabolism SLC (AAM-SLC), and Nucleotide Metabolism SLC (NM-SLC). Further analysis revealed that osteoclasts had relatively higher CM-SLC and NM-SLC scores, myeloid cells showed elevated AAM-SLC scores, and tumor cells had NM-SLC scores second only to osteoclasts (Fig,1 A, Fig. 2D- E). In contrast, LM-SLC scores were consistently low across different cell types (Fig. 2F).
Fig. 2
Landscape of SLCs in the scRNA-seq dataset for osteosarcoma. (A) Heatmap showing the expression of SLCs in each cell type. (B) Differential expression analysis of SLCs across each cell types. An adjusted p value < 0.01 is indicated in red, while an adjusted p value ≥ 0.01 is indicated in black. (C) Ridge plot of SLC score in each cell type. (D) t-SNE plot of SLC score in osteosarcoma dataset. (E) Enrichment score of Carbohydrates Metabolism SLC; CM-SLC Score in each cell type (left), t-SNE of CM-SLC Score(right). (F) Enrichment score of Lipid Metabolism SLC; LM-SLC Score in each cell type (left), t-SNE of LM-SLC Score(right). (G) Enrichment score of Amino Acid Metabolism SLC; AAM-SLC Score in each cell type (left), t-SNE of AAM-SLC Score(right). (H) Enrichment score of Nucleotide Metabolism SLC; NM-SLC Score in each cell type (left), t-SNE of NM-SLC Score(right)
Construction of a prognostic risk scoring model for osteosarcoma based on SLCs and survival analysisWe conducted a consensus clustering analysis based on 449 solute carrier (SLC) genes within osteosarcoma cases from the TARGET database(n = 85). This analysis identified two distinct clusters (K = 2, Cluster A = 41, Cluster B = 44) as determined by the consensus matrix, area under the curve, and cumulative distribution function (CDF) (Fig S1A- D). Survival analysis indicated that patients in Cluster B had a worse prognosis than those in Cluster A (p = 0.037) (Fig S1E). A clustering heatmap illustrated the correlation of SLC family gene expression with clinical features such as gender, age, ethnicity, and tumor site, revealing no significant inter-cluster differences in these characteristics (Fig S1F, Supplementary Table 3).
To further investigate the role of the SLC family in osteosarcoma progression and clinical prognosis, we randomly divided the TARGET osteosarcoma cohort into a training group (n = 50) and a testing group (n = 35) in a 6:4 ratio. Subsequently, we employed LASSO penalized Cox regression analysis to identify SLC family genes associated with prognosis, selecting the penalty parameter based on the minimum criterion (Fig. 3A, B). This analysis identified nine prognostic genes, among which SLC7A1 had the highest risk ratio of 2.19 (p = 0.011) (Fig. 3C). Based on the median risk score, we classified the training set into high-risk and low-risk groups. Survival analysis demonstrated that the high-risk group had significantly shorter overall survival compared to the low-risk group. The prognostic model exhibited high sensitivity and specificity, with area under the receiver operating characteristic (ROC) curve values of 0.856 for 1-year survival prediction, 0.961 for 3-year survival prediction, and 0.964 for 5-year survival prediction (Fig. 3D, E). Similar results were observed in the testing set and the external validation cohort GSE21257 (Fig. 3F- I). Further survival analysis of the genes identified through LASSO regression revealed that NIPA2 and SLC45A4 expression levels were positively correlated with prognosis, indicating that high-expression groups had better outcomes; In contrast, expressions of SLC13A5, SLC38A5, and SLC7A1 were negatively correlated with prognosis (Fig S2A- I).
Fig. 3
Construction of an SLCs-related prognostic signature in osteosarcoma. (A) Coefficient profiles of SLCs. (B) Identification of the best parameter(lambda) in LASSO. (C) Cox analysis of identified 9 genes to construct the risk model. (D) Kaplan-Meier analysis of overall survival in training group. (E) Receiver operating characteristic (ROC) curve analysis for 1-,3-, and 5-year survival for risk model in training group. (F) Kaplan-Meier analysis of overall survival in testing group. (G) ROC curve analysis for 1-,3-, and 5-year survival for risk model in testing group. (H) Kaplan-Meier analysis of overall survival in GSE21257 cohort. (I) ROC curve analysis for 1-,3-, and 5-year survival for risk model in GSE21257 cohort. (J) Nomogram for predicting 1-, 3-, and 5-year survival rate in osteosarcoma. (K) Calibration curves for internal validation of the nomogram. (L) ROC curve analysis of the nomogram in predicting the overall survival
Subsequently, we constructed a nomogram that incorporated clinical parameters alongside the SLC risk model to predict overall survival in the TARGET-OS cohort (Fig. 3J). The internal validation calibration plot of the nomogram demonstrated a strong correlation between predicted probabilities and actual observed outcomes (Fig. 3K). The combined nomogram achieved an AUC value of 0.848, indicating good accuracy in predicting overall survival rates (Fig. 3L).
Immune landscape of SLC-related prognostic signature in osteosarcoma patientsWe conducted a differential analysis of RNA sequencing data across different risk groups in the TARGET-OS cohort. Using the online tool Metascape, we performed Gene Ontology (GO) enrichment analysis on differential genes (|logFC| > 1, p < 0.05) and constructed an enrichment network. The results revealed significant enrichment of differential genes in pathways related to homophilic cell adhesion through plasma membrane adhesion molecules, immunoglobulin-mediated immune response, and activation of immune responses (Fig. 4A, B).
Fig. 4
Immune landscape of SLC-related prognostic signature in osteosarcoma patients. (A) GO annotation of differential genes between high and low-risk groups. (B) Network of enriched terms. (C) Comparison of immune score, stromal score, estimate score, and tumor purity between low- and high-risk groups. (D) Immune cell fraction analysis by CIBERSORT. (E) Immune cell infiltration proportions between low- and high-risk groups by CIBERSORT. (F) Immune cell infiltration proportions between low- and high-risk groups by XCELL and MCPcounter. (G) Expression of immune checkpoint genes between low- and high-risk groups
The ESTIMATE algorithm assessed the infiltration of stromal and immune cells in the TARGET-OS cohort, indicating that the high-risk group exhibited lower immune scores, stromal scores, and ESTIMATE scores, alongside a higher tumor purity (Fig. 4C). Further analysis using the CIBERSORT algorithm evaluated the composition of immune cells in each sample (Fig. 4D), revealing differences in the infiltration of naive CD4 T cells and memory-activated CD4 + T cells between high- and low-risk groups (Fig. 4E). Additionally, the XCELL and MCPcounter algorithms indicated significant differences in the infiltration levels of macrophages, mast cells, and monocytes between the two groups (Fig. 4F).
Differential expression analysis of immune checkpoint genes revealed that the expression levels of CD274, CD44, CTLA4, ICOS, KIR3DL1, LAG3, NPR1, TIGIT, and TNFSF4 were significantly higher in the low-risk group compared to the high-risk group (Fig. 4G). We also examined the correlation between six SLC family genes identified as significantly associated with prognosis in the risk model and immune cell infiltration. Notably, these genes correlated significantly with the infiltration of multiple immune cell types; specifically, SLC7A1 showed a significant negative correlation with activated NK cells and monocytes, while demonstrating a positive correlation with activated dendritic cells (DCs) (Fig. 4H). These findings suggest that the expression of SLC family genes is associated with immunomodulation in osteosarcoma, highlighting their potential regulatory role in the immune microenvironment.
Single-cell analysis of SLC7A1 in osteosarcoma cellsGiven that SLC7A1 demonstrated the highest risk ratio in the TARGET-OS cohort and is negatively correlated with prognosis, we further analyzed it using osteosarcoma single-cell data. The bubble plot revealed elevated expression levels of SLC7A1 and a higher proportion of expressing cells among the OS cells (Fig. 5A). Further clustering analysis categorized OS cells into five distinct groups: OS1, OS2, OS3, OS4, and OS5, each with unique gene expression profiles (Fig. 5B, C). Notably, SLC7A1-positive cells were predominantly found in the OS4 group (Fig. 5D- F).
Fig. 5
Single-cell analysis of SLC7A1 in osteosarcoma cells. (A) Average and percent expression of SLC7A1 in each cell types. (B) t-SNE plot showing the clustering of OS cells. (C) Heatmap showing the differential genes in each OS Clusters. (D) Average and percent expression of SLC7A1 in each OS Clusters. (E) t-SNE plot of SLC7A1 expression in OS cells. (F) t-SNE plot showing density of SLC7A1 in OS cells. (G) Pseudotemporal ordering trajectory map of OS cells. (H) Pseudotemporal ordering of OS Clusters. (I) Trajectory plot showing the expression of SLC7A1. (J) SLC7A1 pseudotemporal expression map. (K) scMetabolism analysis of OS Clusters. (L) The tumor Hallmark scores of SLC7A1-positive and SLC7A1-negative cells in OS cells
Using the Monocle algorithm to infer the differentiation trajectory of OS cells (Fig. 5G), we determined that the OS4 group cells occupy the terminal end of this trajectory (Fig. 5H), with SLC7A1 expression differing significantly along it (p<0.001) (Fig. 5I, J). We assessed the metabolic pathway activity across tumor groups using the scMetabolism algorithm and found that, consistent with high SLC7A1 expression, OS4 cells exhibited enhanced activity in pathways such as arginine biosynthesis, arginine and proline metabolism, and glycolysis/gluconeogenesis (Fig. 5K). Hanahan et al. identified 14 hallmarks of cancer, including “limitless replicative potential,” “tissue invasion and metastasis,” and “tumor-promoting inflammation,” which reflect the potential for malignant progression [34]. Based on these hallmarks, we retrieved the corresponding gene sets to evaluate the malignancy of the tumor cells [35]. Scoring of 14 tumor characteristic gene sets revealed that SLC7A1-positive cells possess greater proliferative and metastatic capabilities, as well as enhanced stemness (Fig. 5L).
SLC7A1 promotes the proliferation and metastasis of osteosarcoma in vitro and correlates with poor prognosisImmunoblotting revealed that SLC7A1 is upregulated in tumor tissues (Fig. 6A). Subsequently, we selected the 143B and SJSA-1 osteosarcoma cell lines and performed SLC7A1 knockdown in vitro (Fig. 6B). This knockdown resulted in reduced proliferative capacity (Fig. 6C), which was also indicated by a lower proportion of positive EdU cells and fewer colony formations (Fig. 6D, E). Furthermore, SLC7A1 knockdown significantly inhibited cell invasion, migration, and sphere-forming abilities in both cell lines (Fig. 6F- H). To validate the prognostic impact of SLC7A1, we analyzed its expression in 70 osteosarcoma specimens using immunohistochemistry (IHC) (Fig. 6I). Results indicated that 53% of patients exhibited strong or moderate SLC7A1 staining, while 47% showed weak or negative staining. Kaplan-Meier analysis revealed that high SLC7A1 expression correlated with poorer overall survival (OS) (p = 0.0048) and lung metastasis-free survival (LMFS) (p = 0.0017) (Fig. 6J). Both univariate and multivariate regression analyses identified SLC7A1 levels as an independent prognostic factor (Fig. 6K). These findings suggest that SLC7A1 may be a valuable therapeutic target and prognostic marker in osteosarcoma.
Fig. 6
SLC7A1 promotes the proliferation and metastasis of osteosarcoma in vitro and is correlates with poor prognosis. (A) SLC7A1 protein levels measured in 6 matched pairs of osteosarcoma and adjacent normal tissues by immunoblotting. (B) Knockdown of SLC7A1 in 143B and SJSJ1 cell lines. (C) Proliferation assay of SLC7A1-KD 143B (left) and SJSA-1 (right) cells (n = 6). (D) Colony formation of SLC7A1-KD 143B and SJSA-1 cells (left). Quantification of colony formation (right) (n = 3). (E) EdU incorporation assay of SLC7A1-KD 143B and SJSA-1 cells (left). Quantification of EdU incorporation assay (right). Scale bar: 100 μm (n = 3). (F) Migration and invasion of SLC7A1-KD 143B cells (left). Quantification of migration and invasion (right). Scale bar: 100 μm (n = 3). (G) Migration and invasion of SLC7A1-KD SJSA-1 cells (left). Quantification of migration and invasion (right). Scale bar: 100 μm (n = 3). (H) Sphere formation assay of SLC7A1-KD 143B and SJSA-1 cells (left). Quantification of Sphere formation assay (right) (n = 3). (I) Representative IHC images showing high or low expression of SLC7A1 in patient tumor specimens. Scale bars: 50 μm (left), 25 μm (right). (J) Overall survival (left) and LMFS (right) of patients according to the expression levels of SLC7A1, by log-rank test. (K) Univariate and multivariate analyses of prognostic factors for overall survival and LMFS among patients with osteosarcoma, by Wald test. Data are presented as the mean ± SD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, by two-way ANOVA with Dunnett’s multiple comparisons test (C) and one-way ANOVA with Dunnett’s multiple comparisons test (D, E, F, G, and H)
Relationship between SLC7A1 expression and tumor immune microenvironmentTo investigate the relationship between SLC7A1 expression in tumor cells and the tumor immune microenvironment, we categorized 15 single-cell samples into high and low SLC7A1 expression groups based on the levels of SLC7A1 in the tumor cells. The analysis revealed distinct cell type compositions, with a notable decrease in myeloid cells in the high-expression group (Fig. 7A). Further dimensionality reduction and clustering of isolated myeloid cells identified four distinct types: tumor-associated macrophages (TAMs), dendritic cells, monocytes, and proliferative myeloid cells, each with unique expression profiles (Fig. 7B- D). A re-analysis of cellular composition confirmed a significant reduction in TAMs in the high-expression group (Fig. 7E).
Fig. 7
Analysis of immune cell proportions and cell communication in SLC7A1-associated groups (A) Cell proportion in different SLC7A1 expression groups. (B) Clustering of myeloid cells. (C) Feature plots of marker genes in each clusters of myeloid cells. (D) Bubble plot of the average and percent expression of different markers in each clusters of myeloid cells. (E) Cell proportion of TAMs in different SLC7A1 expression groups. (F) scMetabolism analysis of TAMs in different SLC7A1 expression groups. (G) Heatmap of cell-cell communication between OS cells and immune cells. Number of interactions (left); Strength of interactions (right). (H) Network of cell-cell communication between OS cells and immune cells. Number of interactions (left); Strength of interactions (right). (I) Heatmap showing differential interactions between OS cells and immune cells. (J) Differential cell-cell communication signaling pathways between OS cells and TAMs in different SLC7A1 expression groups. (K) Bubble heatmap showing upregulated receptor-ligand interactions between OS cells and TAMs in High-SLC7A1 group. (L) Chord diagram showing upregulated receptor-ligand interactions between OS cells and TAMs in High-SLC7A1 group
Utilizing the scmetabolism package, we conducted metabolic analyses on TAMs and found that the high SLC7A1 expression group exhibited enhanced arginine biosynthesis, while arginine and proline metabolism was diminished, suggesting a potential arginine deficiency in this microenvironment (Fig. 7F). Additionally, using CellChat, we explored ligand-receptor interactions between tumor cells and various immune cells, finding a higher frequency and intensity of interactions between tumor cells and macrophages (Fig. 7G). TAMs received more signals from osteosarcoma cells compared to other immune cells (Fig. 7H).
Further comparisons of intercellular communication between the high and low SLC7A1 expression groups indicated that osteosarcoma cells in the high SLC7A1 expression group sent more signals to TAMs, albeit with slightly reduced signal intensity (Fig. 7I). Differential signal pathway analysis revealed enhanced signaling in the high SLC7A1 expression group for pathways such as ANGPTL, THBS, and IFN-I, whereas the low SLC7A1 expression group exhibited stronger signaling in MHC-I, CD86, and CD80 (Fig. 7J). Notably, pathways including THBS1-CD47, SPP1-CD44, PTN-SDC3, and IL6-(IL6R + IL6ST) were significantly upregulated in the high SLC7A1 expression group (Fig. 7K).
SLC7A1 influenced M2 macrophage polarizationTo further clarify the regulatory role of SLC7A1 on macrophages, we examined the effect of SLC7A1 expression in osteosarcoma cells on the differentiation of tumor-associated macrophages (TAMs) using a co-culture model (Fig. 8A). THP1 cells were treated with PMA to induce macrophage differentiation and then co-cultured with SLC7A1-knockdown osteosarcoma cells (143B). Following this, THP1 cells were collected for analysis. Flow cytometry revealed that the SLC7A1-knockdown group exhibited a significant decrease in CD206 expression and a slight increase in CD86 expression compared to control cells (Fig. 8B, C). Additionally, qPCR analysis demonstrated significant upregulation of M2 macrophage markers, including CD206, ARG1, IL-10, TGFB1, and CD163 (Fig. 8D). These findings indicate that SLC7A1 expression in osteosarcoma cells facilitates the polarization of M2-like macrophages within the microenvironment.
Fig. 8
SLC7A1 influenced M2 macrophage polarization. (A) Schematic diagram of the co-culture model workflow between osteosarcoma cells and macrophages derived from induced THP1 cells. (B) CD206 expression in TAMs co-cultured with control or SLC7A1-KD 143B cells, detected by flow cytometry (left). Quantification of CD206 + TAMs (right) (n = 3). (C) CD86 expression in TAMs co-cultured with control or SLC7A1-KD 143B cells, detected by flow cytometry (left). Quantification of CD86 + TAMs (right) (n = 3). (D) The expression of CD206, ARG1, IL10, TGFB1, and CD163 mRNA in TAMs co-cultured with control or SLC7A1-KD 143B cells, detected by qRT-PCR analyses (n = 3). Data are presented as the mean ± SD, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, by Students’ t-test (B, C, and D)
Structure-based virtual screen identified Cepharanthine as a potential inhibitor of SLC7A1Structural-based virtual screening was conducted to identify approved drugs that may inhibit SLC7A1 function (Fig. 9A). We utilized the ZINC20 database, which contains nearly two billion downloadable compound structures, to retrieve and download the structural files of 5,903 approved drugs, converting them into 3D models. The SLC7A1 protein structure was predicted using AlphaFold2 (Fig. 9B), with a predicted alignment error plot indicating high confidence in most segments (Fig. 9C). Model quality assessment via the ProSA server yielded a ProSA Z-score of -6.78, indicating that the model’s quality is comparable to those derived from NMR and X-ray crystallography (Fig. 9D). Virtual screening was performed using AUTODOCK VINA, designating SLC7A1 as the receptor and the 5,903 approved drugs as ligands, from which the top 10 drugs with the lowest docking scores were selected (Table 1). The molecular docking visualization of these drugs is depicted, showing the SLC7A1 protein model in white, the drug molecules in blue sticks, hydrogen bonds as yellow dashed lines, and labeled amino acid residues involved in interactions (Fig. 9E- N).
Fig. 9
Structure-based virtual screen of SLC7A1 (A) Schematic diagram of the workflow for structure-based virtual screening. (B) The predicted 3D structure of SLC7A1 by AlphaFold2. (C) Predicted aligned error of the SLC7A1 3D model. (D) Overall model quality of the SLC7A1 3D analyzed by ProSA-Web. (E) The calculated binding mode between Paritaprevie and SLC7A1. (F) The calculated binding mode between Cepharanthine and SLC7A1. (G) The calculated binding mode between Midostaurin and SLC7A1. (H) The calculated binding mode between Ledipasvir and SLC7A1. (I) The calculated binding mode between Alcuronium and SLC7A1. (J) The calculated binding mode between Venetoclax and SLC7A1. (K) The calculated binding mode between Eltrombopag and SLC7A1. (L) The calculated binding mode between Simeprevir and SLC7A1. (M) The calculated binding mode between Vaprisol and SLC7A1. (N) The calculated binding mode between Glecaprevir and SLC7A1
To further screen these 10 drugs, we first assessed their impact on arginine uptake in osteosarcoma cells. The results revealed that at a concentration of 10 µM, only Paritaprevir, Cepharanthine, Eltrombopag, and Simeprevir significantly inhibited arginine uptake (Fig. 10A). IC50 analysis indicated that Cepharanthine (CPE) (5.13 µM) had the lowest half-inhibitory concentration against osteosarcoma cells (Fig. 10B). Cellular Thermal Shift Assay (CETSA) demonstrated that CPE inhibited heat-induced degradation of SLC7A1 protein (Fig. 10C), suggesting a potential interaction between CPE and SLC7A1. Treatment of osteosarcoma cells with CPE at concentrations of 0, 2, and 5 µM revealed that, with increasing concentration, CPE enhanced its inhibitory effects on both arginine uptake and cell proliferation (Fig. 10D, E). To evaluate the effect of CPE pre-treatment on osteosarcoma cell-induced macrophage polarization, we collected conditioned media from osteosarcoma cells pretreated with or without CPE for 24 h and used it to culture THP1-derived macrophages (Fig. 10F). After 48 h of incubation, flow cytometry analysis revealed that CPE pretreatment significantly downregulated CD206 expression and upregulated CD86 expression, with these effects intensifying as CPE concentration increased (Fig. 10G, H). These findings indicate that Cepharanthine exerts anti-tumor effects by binding to SLC7A1 and inhibiting its activity.
Fig. 10
Cepharanthine is a potential targeted inhibitor of SLC7A1. (A) Inhibition of arginine uptake in 143B cells by 10 drugs. (B) IC50 analysis of Paritaprevir, Cepharanthine, Eltrombopag, and Simeprevir. (C) CETSA showing the binding of CPE to SLC7A1 protein. (D) Arginine uptake in 143B cells treated with 0, 2, and 5 µM CPE. (E) CCK8 assay evaluating the proliferation of 143B cells treated with 0, 2, and 5 µM CPE. (F) Schematic of conditioned medium production from CPE -pretreated 143B cells. Cells were treated with 0, 2, or 5 µM CPE, followed by medium replacement after 24 h. After an additional 24-hour incubation, the conditioned medium was collected. This medium was used to culture THP1-induced macrophages for 48 h before flow cytometry analysis. (G) Flow cytometry analysis of CD206 expression in macrophages. (H) Flow cytometry analysis of CD86 expression in macrophages
Table 1 10 drugs with the lowest docking scores
留言 (0)