
記住我
Noonan syndrome with multiple lentigines (NSML), previously known as LEOPARD syndrome, is a rare multisystem autosomal dominant disorder first described in 1969. It is characterized by lentigines, electrocardiographic conduction abnormalities, hypertelorism, pulmonary stenosis, genital anomalies, growth retardation, and deafness (1, 2). Approximately half of the cases are due to variants in the PTPN11 gene, affecting the RAS-MAPK signaling pathway, which influences cell proliferation, migration, and differentiation (3). Around 85% of patients have cardiac defects, most commonly HCM and pulmonary stenosis, with occasional aortic and mitral valve anomalies (4). The electrocardiogram (ECG) shows not only abnormalities associated with hypertrophic cardiomyopathy but also conduction abnormalities (4). However, the clinical features and genetic heterogeneity of this condition vary significantly among patients (3).
Case presentationA 58-year-old female presented with a 10-year history of exertional dyspnea, progressively worsening bilateral lower extremity edema, and new-onset orthopnea. She had a history of congenital bilateral hearing loss. Diffuse brown pigmented macules appeared on the face, trunk, and limbs since childhood, with clear borders and varying sizes and shapes. Physical examination revealed short stature (height: 158.5 cm), high forehead hairline, hypertelorism, ptosis, low-set ears, short neck, dental malalignment, and pectus excavatum. Left-sided talipes equinovarus was also noted (Figure 1). Her intellectual development was normal. Cardiac examination showed an enlarged heart, irregular rhythm, and grade 3/6 systolic murmurs at the mitral and tricuspid areas. Jugular venous distention and significant bilateral lower extremity edema were also found. The patient had eight siblings. Through telephone communication and photographic evidence, none of them had a history of heart disease, generalized macules, or developmental anomalies. Her first husband was intellectually disabled, and they had a son who was also intellectually disabled with multiple lentigines. With her second husband, she had a daughter who was physically and intellectually normal and had no lentigines.
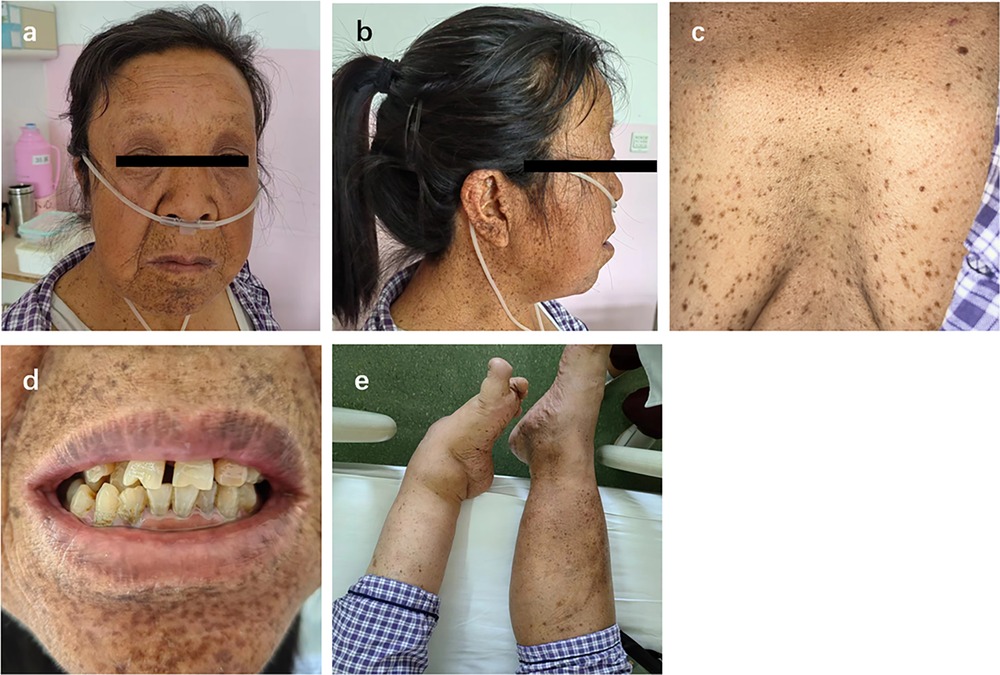
Figure 1. Clinical features of the patient. (a,b) Facial characteristics; (c) Lentigines and pectus excavatum; (d) Facial lentigines and dental malalignment; (e) Left foot deformity.
Laboratory tests showed brain natriuretic peptide (BNP) 845 pg/ml and high-sensitivity cardiac troponin I (hs-cTNI) 9.0 ng/L. Echocardiography (UCG) and chest x-ray (Figure 2) revealed global cardiac enlargement (left atrium: anteroposterior diameter 6.4 cm, superior-inferior diameter 8.9 cm, transverse diameter 5.9 cm; right atrium: superior-inferior diameter 9.2 cm, transverse diameter 6.6 cm with an interatrial membrane; left ventricle: end-diastolic diameter 5.8 cm, end-systolic diameter 3.3 cm, posterior wall thickness 1.1 cm, interventricular septum 1.0 cm; right ventricle: anteroposterior diameter 2.4 cm, transverse diameter 5.1 cm), left ventricular ejection fraction 73.7%, severe mitral and tricuspid regurgitation, and an inferior vena cava diameter of 3.6 cm. Septal defect or pulmonary artery stenosis was not found. ECG indicated atrial fibrillation with a ventricular rate of 117 bpm (Figure 3). 1,530 premature ventricular contractions (PVCs) of various morphologies and 4 short runs of ventricular tachycardia were recorded by Holter ECG throughout the day (Figure 3). Furthermore, the patient reported no history of alcohol, drug, or tobacco use, and there was no diagnosis of hypertension. While coronary imaging could not be performed due to the patient's personal preference, bilateral carotid artery ultrasound revealed no atherosclerotic plaques, suggesting a low likelihood of coronary artery disease as a contributing factor. Whole exome sequencing was performed due to suspicion of NSML, revealing a heterozygous pathogenic variant in PTPN11, exon 12, c.1403 C>T (p.Tyr468Met) in the blood samples of both the patient and her son (Figure 4). However, this mutation was not detected in her daughter's blood sample (Figure 4). Genetic testing was not performed on her siblings. Other pathogenic genetic alterations related to cardiomyopathy and arrhythmias were not found to have significant relevance, especially genes for the Rasopathies and the cardiovascular phenotype based on the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines (5). The patient was treated with oral anticoagulants, beta-blockers, and diuretics. Symptoms improved, and the patient was discharged with a follow-up plan. The first follow-up visit was conducted after 15 months. The patient's heart rate is well-controlled, with a mean ventricular rate of 79 beats per minute on Holter monitoring, which indicated 24 h of atrial fibrillation and 26 premature ventricular contractions. The left ventricular end-diastolic diameter remained unchanged at 5.6 cm, leading us to conclude that the likelihood of tachycardia-induced cardiomyopathy is low.
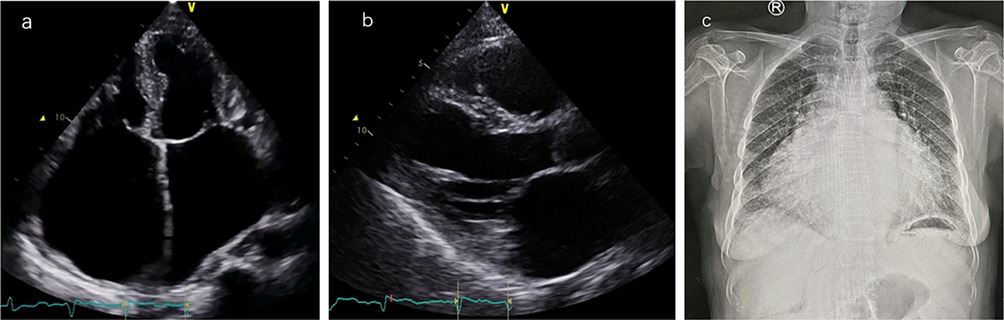
Figure 2. Cardiac imaging findings. (a) Apical four-chamber view: the left atrium has a vertical diameter of 8.9 cm and a transverse diameter of 5.9 cm; the right atrium has a vertical diameter of 9.2 cm and a transverse diameter of 6.6 cm. (b) Left ventricular long-axis view: left ventricular end-diastolic diameter (LVEDD) is 5.8 cm, left ventricular posterior wall thickness (LVPWT) is 1.1 cm, interventricular septal thickness (IVS) is 1.0 cm, and right ventricular anteroposterior diameter is 2.4 cm. (c) Anteroposterior chest x-ray showed significant heart enlargement.

Figure 3. Electrocardiographic findings. (a) ECG on admission: atrial fibrillation. (b) Holter monitoring: multiple premature ventricular contractions of varying morphologies, some occurring in pairs, and short runs of ventricular tachycardia.

Figure 4. Sanger sequencing of the PTPN11 variant, exon 12, c.1403 C>T (p.Tyr468Met). (a) Proband: Heterozygous mutation detected at the indicated position (red arrow). (b) Proband's son: Heterozygous mutation detected at the same position (red arrow). (c) Proband's daughter: No mutation detected at this position.
DiscussionIn this case report, we highlight the cardiac manifestations of a patient with NSML. Unlike most NSML patients, this patient did not exhibit common cardiovascular manifestations such as hypertrophic cardiomyopathy or pulmonary stenosis. Instead, the primary manifestations were whole cardiac enlargement, heart failure with preserved ejection fraction, and atrial fibrillation. As far as we know, previous NSML cases have not reported such a cardiac phenotype with whole cardiac enlargement (4, 6–8). This case contributes to a broader understanding of the cardiac manifestations of NSML.
NSML is known as Leopard syndrome previously and is classified as a RASopathy, which refers to a group of related disorders caused by variants in genes within the Ras/mitogen-activated protein kinase (Ras/MAPK) pathway (9). The prevalence of NSML is unknown, but it is considered a rarer phenotype among RASopathies. Multiple gene variants can cause Noonan Syndrome/NSML, including PTPN11, BRAS, and RAF1, with PTPN11 variants being the most common one. Variants in exon 7 and exon 12 of the PTPN11 gene, particularly the Tyr279Cys and Thr468Met varitants, are most frequent (10). The cardiac phenotype of patients may be related to specific exon variants. For example, variants in exon 7 and exons 12 and 13 are associated with the septal morphology of hypertrophic cardiomyopathy (11). The protein product of PTPN11, the protein tyrosine phosphatase (SHP2), plays a clear role in valve morphogenesis. In mouse models, cardiac defects are caused by the activation of mutant SHP2 in the developing endocardium, which further affects downstream signaling (12). Histologically, myocardial tissues from PTPN11 mutant mice showed hypertrophy of myocardial cells, disarrayed myocardial fibers, and infiltration of inflammatory cells in the intercellular spaces. As the mice aged to 52 weeks, significant left ventricular hypertrophy transitioned to a dilated cardiomyopathy phenotype, characterized by cardiac enlargement, thinning of the ventricular walls, and impaired cardiac contractility (13) Currently, there is no clear experimental evidence to explain the molecular mechanism underlying the whole cardiac enlargement observed in NSML patients. In one NSML family, elderly patients did not show a cardiac enlargement phenotype (14), but variants in exon 7 of PTPN11 primarily caused the pathogenic gene in this family. There may still be differences in clinical phenotypes and disease progression among NSML patients with different variants. Besides, current diagnostic tools cannot entirely rule out coronary artery disease or unknown genetic variants that may be contributing to the patient's cardiac enlargement.
In terms of electrophysiological phenotype, multifocal and ectopic atrial tachycardia has been reported in pediatric patients with RASopathies, and some patients respond well to flecainide treatment, suggesting that these patients may have dysregulated calcium ion release, which could lead to cardiomyopathy and heart failure (15). More common ECG findings include ventricular hypertrophy, Q waves, prolonged QT intervals, and repolarization abnormalities (4). There have also been reports of NSML patients exhibiting non-sustained ventricular tachycardia, and it seems that patients without PTPN11 variants do not have an increased risk of arrhythmia. These patients also have left atrial enlargement, which may be related to diastolic dysfunction or outflow tract obstruction (16). There is no experimental evidence to clarify the mechanism of arrhythmia in NSML patients. We hypothesize that the atrial fibrillation and polymorphic PVCs in this patient may be secondary to cardiac structural changes caused by whole cardiac enlargement.
ConclusionWe report a patient with distinctive facial features and multiple lentigines who was diagnosed with NSML through genetic testing. The heart is a commonly affected organ in NSML, but the manifestation of whole cardiac enlargement is rare. This case is significant as it emphasizes the importance of recognizing the various cardiac manifestations in NSML, especially in patients with exon 12 of the PTPN11 mutation, and highlights the need to consider differential diagnoses in heart failure patients with distinctive facial features.
Data availability statementThe raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statementThe studies involving humans were approved by Biomedical Research Ethics Committee, Peking University First Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by- product of routine care or industry. Written informed consent for participation was not required from the participants or the participants' legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributionsLF: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. JJ: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. YZ: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. XH: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. WD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. XX: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – review & editing. YJ: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
AcknowledgmentsThe authors would like to thank the patient and her children for their participation in this study.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References4. Limongelli G, Pacileo G, Marino B, Digilio MC, Sarkozy A, Elliott P, et al. Prevalence and Clinical Significance of Cardiovascular Abnormalities in Patients With the LEOPARD Syndrome. Am J Cardiol. (2007) 100(4):736–41. doi: 10.1016/j.amjcard.2007.03.093
PubMed Abstract | Crossref Full Text | Google Scholar
5. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
PubMed Abstract | Crossref Full Text | Google Scholar
8. Baldo F, Fachin A, Da Re B, Rubinato E, Bobbo M, Barbi E. New insights on Noonan syndrome’s clinical phenotype: a single center retrospective study. BMC Pediatr. (2022) 22(1):734. doi: 10.1186/s12887-022-03804-2
PubMed Abstract | Crossref Full Text | Google Scholar
10. Sarkozy A, Conti E, Seripa D, Digilio MC, Grifone N, Tandoi C, et al. Correlation between PTPN11 gene mutations and congenital heart defects in Noonan and LEOPARD syndromes. J Med Genet. (2003) 40(9):704–8. doi: 10.1136/jmg.40.9.704
PubMed Abstract | Crossref Full Text | Google Scholar
11. Kauffman H, Ahrens-Nicklas RC, Calderon-Anyosa RJC, Ritter AL, Lin KY, Rossano JW, et al. Genotype-phenotype association by echocardiography offers incremental value in patients with Noonan syndrome with multiple lentigines. Pediatr Res. (2021) 90(2):444–51. doi: 10.1038/s41390-020-01292-7
PubMed Abstract | Crossref Full Text | Google Scholar
12. Araki T, Chan G, Newbigging S, Morikawa L, Bronson RT, Neel BG. Noonan syndrome cardiac defects are caused by PTPN11 acting in endocardium to enhance endocardial-mesenchymal transformation. Proc Natl Acad Sci U S A. (2009) 106(12):4736–41. doi: 10.1073/pnas.0810053106
PubMed Abstract | Crossref Full Text | Google Scholar
13. Marin TM, Keith K, Davies B, Conner DA, Guha P, Kalaitzidis D, et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome–associated PTPN11 mutation. J Clin Invest. (2011) 121(3):1026–43. doi: 10.1172/JCI44972
PubMed Abstract | Crossref Full Text | Google Scholar
14. Chan C-H, Chu M-F, Lam UP, Mok T-M, Tam W-C, Tomlinson B, et al. Case report: Distinctive cardiac features and phenotypic characteristics of Noonan syndrome with multiple lentigines among three generations in one family. Front Cardiovasc Med. (2023) 10:1225667. doi: 10.3389/fcvm.2023.1225667
PubMed Abstract | Crossref Full Text | Google Scholar
15. Levin MD, Saitta SC, Gripp KW, Wenger TL, Ganesh J, Kalish JM, et al. Nonreentrant atrial tachycardia occurs independently of hypertrophic cardiomyopathy in RASopathy patients. Am J Med Genet A. (2018) 176(8):1711–22. doi: 10.1002/ajmg.a.38854
PubMed Abstract | Crossref Full Text | Google Scholar
16. Limongelli G, Sarkozy A, Pacileo G, Calabrò P, Digilio MC, Maddaloni V, et al. Genotype–phenotype analysis and natural history of left ventricular hypertrophy in LEOPARD syndrome. Am J Med Genet Part A. (2008) 146A(5):620–8. doi: 10.1002/ajmg.a.32206
留言 (0)