Patients
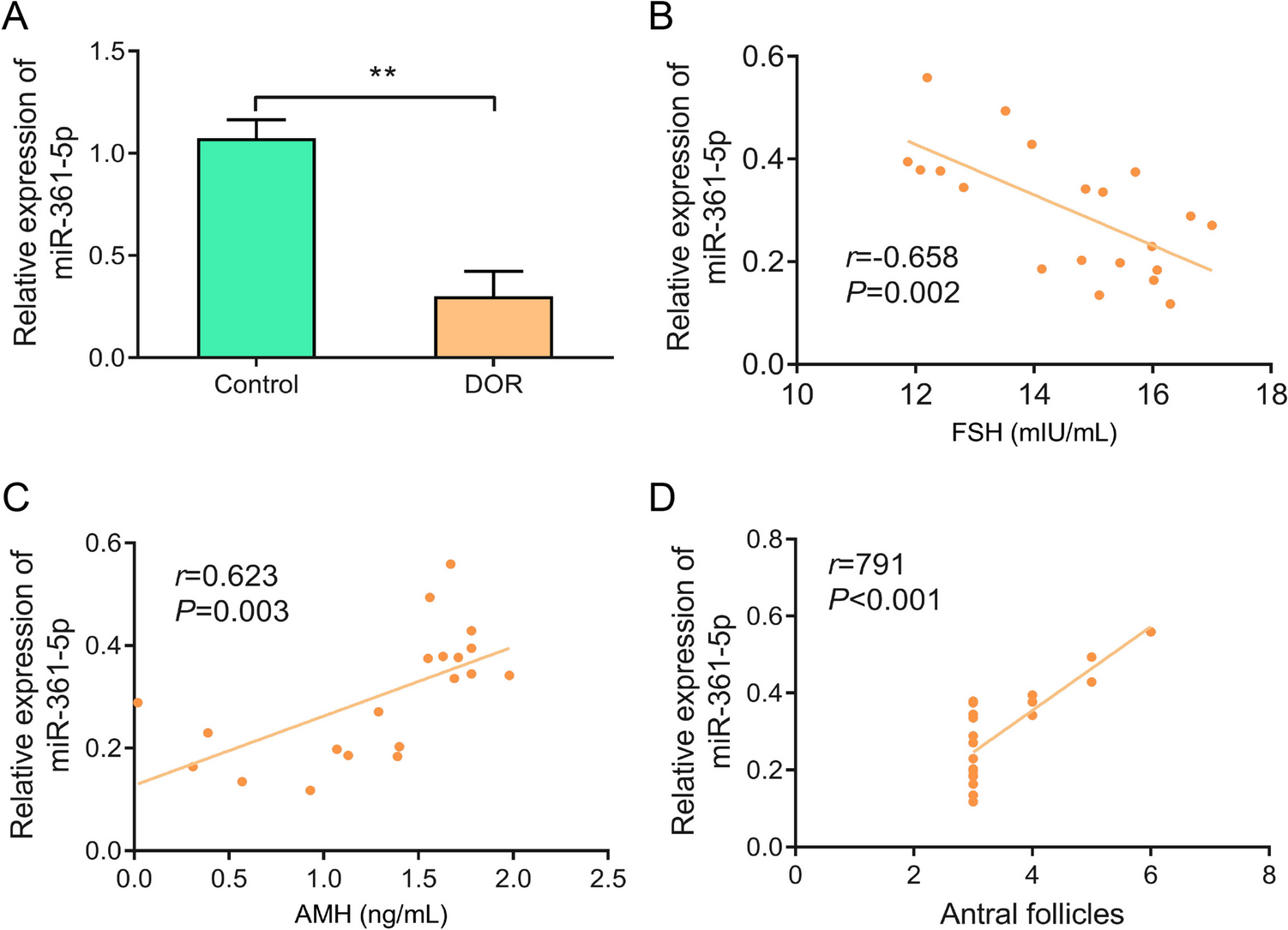
From 2023.2 to 2024.2, patients undergoing assisted reproductive technology treatment at the Reproductive and Genetics Center of our hospital were included in this study. The inclusion criteria were patients aged between 20 and 40 years who had been unable to conceive naturally for over a year. Patients with polycystic ovary syndrome, premature ovarian failure, endometriosis, reproductive endocrine diseases, ovarian tumors, or those who had undergone chemotherapy or pelvic radiotherapy were excluded. A total of 20 patients were diagnosed with DOR, while 20 patients with normal ovarian function served as the control group. The control group patients were infertile due to tubal or male factors. Follicular fluid was collected from all patients. Clinical information collected included age, BMI, duration of infertility (years), follicle-stimulating hormone (FSH, mIU/mL), luteinizing hormone (LH, mIU/mL), FSH/LH ratio, estradiol (E₂, pg/mL), progesterone (ng/mL), anti-Müllerian hormone (AMH, ng/mL), and antral follicle count. The study protocol was approved by the Institutional Review Board of our hospital. Informed consent was obtained from all patients participating in the study.
Cell culture
KGN cells, a human ovarian granulosa cell line, were used in the study for further experiments. Human granulosa cells were isolated from the follicular fluid of patients undergoing assisted reproductive technology at our hospital. The follicular fluid was collected during oocyte retrieval, and granulosa cells were separated using a density gradient centrifugation method and identified as described previously [21]. After isolation, granulosa cells were immediately cultured in culture medium which was supplemented with 10% fetal bovine serum (Gibco, Australia), 1% Penicillin mixture (Solarbio, Beijing, China), and placed in 5% CO2, 37 °C incubator (Thermo, USA) prior to experimentation. The 3rd generation of cultured cells was selected for further study.
Cell transfection
To investigate the role of miR-361-5p in granulosa cells, KGN cells were transfected with various miRNA mimics and inhibitors using Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer’s instructions. The cells were grouped into the following transfection conditions: negative control (NC) inhibitor, miR-361-5p inhibitor, NC mimic, miR-361-5p mimic, NC shRNA, SLC25A24 shRNA, and combinations of miR-361-5p inhibitor with either NC shRNA or SLC25A24 shRNA. The negative control used in this study refers to a non-specific inhibitor or mimic that does not target any specific gene, used to account for any non-specific effects. These controls, along with the miRNA mimics and inhibitors, were specifically designed and synthesized by Shanghai IBS Biotech Co., Ltd. Transfection efficiency was confirmed by quantitative real-time PCR (qRT-PCR) and Western blot analysis, ensuring effective modulation of miR-361-5p and SLC25A24 expression.
RT‑qPCR analysis
Total RNA was extracted from cells and exosomes using the TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Reverse transcription and quantitative PCR (qPCR) were performed using the Quant One Step qRT-PCR Kit (Probe) (LM-0102; LMai Biotech Co., Ltd.). Specific primers for miR-25-3p and U6 snRNA (as an internal control) were used to quantify the expression levels. The sequences of the primers synthesized by Tsingke Biotech Ltd. were listed in Table 1.
Table 1 Primer sequence informationWestern blot analysis
Protein extracts from tissues and cells were obtained using the Pierce™ IP Lysis Buffer (Thermo Fisher). Subsequently, 40 μg of protein from each sample was subjected to electrophoresis on a 10% SDS-PAGE gel (Beyotime) and then transferred onto polyvinylidene difluoride (PVDF) membranes (Beyotime). To prevent non-specific binding, the membranes were blocked with 5% fat-free milk for 1 h at room temperature before being incubated with primary antibodies against SLC25A24 (1:1000; Abcam; ab221120), Aromatase (1:2000; Abcam; ab114260), StAR (1:5000; Abcam; ab133657), LC3-II (1:20,000; Abcam; ab103506), LC3-I (1:1000; Abcam; ab192890), BECN1 (1:1000; Abcam; ab207612), P62 (1:1000; Abcam; ab109012), PINK (1:1000; Abcam; ab186303), Parkin (1:2000; Abcam; ab177625), and β-actin (1:5000; Abcam; ab8227) overnight at 4 °C. The following day, the membranes were incubated with a rabbit anti-mouse IgG secondary antibody (1:2000; Abcam) at room temperature for 2 h. β-actin served as the endogenous control throughout the assay.
ELISA
Supernatants from KGN cells and tissues were collected after centrifugation at 1000 g for 10 min. The concentrations of estradiol and progesterone within these supernatants were quantitatively determined employing ELISA kits (Nanjing Jiancheng, China) following the manufacturers’ instructions. Briefly, 100 μL of sample was added to pre-coated plates and incubated for 2 h at room temperature. After washing the plates, a biotinylated detection antibody was added and the plates were incubated. Following a series of washes, streptavidin-HRP was added and the plates were incubated for 30 min. The reaction was developed using TMB substrate and stopped with sulfuric acid.
Intracellular ROS generation detection
The level of intracellular ROS was determined by DCFH-DA staining. Cells were pretreated with indicated drugs for 2 h. Cells were then harvested and incubated with DCFH-DA (10 mM) for 30 min in the dark at 37 °C. After staining, cells were washed twice with D-PBS. The intracellular ROS fluorescence intensity was quantified by flow cytometry and the images were taken by a fluorescence microscope (Olympus IX53; Olympus Corporation, Tokyo, Japan).
Mitochondrial DNA (mtDNA) detection
Quantitative PCR (qPCR) was rigorously applied to assess mtDNA in KGN cells. DNA was extracted meticulously using the Sangon DNA Genome Extraction Kit. For mtDNA analysis, primers mtF3212 and mtR3319 were employed, while for nuclear DNA quantification, the 18S ribosomal RNA gene was targeted with primers 18S1546F and 18S1546R, following the protocol described by Bai et al. [22]. The real-time qPCR was performed on the ABI-Prism 7700 Sequence Detector System. Each 10 μL PCR mixture included 1 × TaqMan Universal PCR Master Mix, 500 nM concentration of each primer, 200 nM TaqMan probe, and a range of 0.2 to 2 ng genomic DNA. The PCR protocol was set to an initial 2 min incubation at 50 °C and 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C for denaturation and 60 s at 60 °C for annealing and extension. This protocol was designed to ensure the precise and reliable quantification of mtDNA.
ATP measurements
Following the protocol described in a previous study [23], ATP levels were measured using a luciferin-luciferase ATP assay system and a CentroPRO LB 962 luminometer, as per the guidelines provided by Molecular Probes (A22066). Briefly, ten denuded oocytes were placed into a 0.2 mL centrifuge tube containing 30 μL of lysis buffer (20 mM Tris, 0.9% Tween 20, and 0.9% Nonidet-40) and homogenized by vortexing until complete lysis was achieved. A standard reaction solution was prepared according to the manufacturer’s instructions and kept on ice away from light until used. For the assay, 5 μL of each sample was transferred into a 96-well plate and equilibrated for 10 s. Subsequently, 150 μL of the reaction solution was added to each well. After a 2-s delay, the luminescence was integrated over 10 s. Light intensity was quantified using ICE software, which is designed for setting up ATP assay protocols. The software’s parameter was preset at 1 for the control samples. The luminescence intensity for the treatment samples was recorded and expressed as relative values compared to the control.
JC-1 staining procedure
Mitochondrial membrane potential was assessed using JC-1 staining (5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide). Cells were seeded in six-well plates and treated according to experimental requirements. Post-treatment, cells were incubated with JC-1 dye (10 μg/mL; Sigma-Aldrich, USA) at 37 °C for 20 min, allowing for adequate staining. Post incubation, cells were washed twice with PBS to remove excess dye. The mitochondrial membrane potential was determined by measuring the intensity of red fluorescence (aggregates, indicating higher potential) and green fluorescence (monomers, indicating lower potential) using a fluorescence microscope.
Dual-luciferase reporter gene assay
Utilizing the bioinformatics tool TargetScan, potential binding sites for miR-361-5p on the 3’-untranslated region (3’-UTR) of SLC25A24 were identified. To experimentally validate the prediction, constructs of luciferase reporter vectors containing the wild-type 3’-UTR of SLC25A24 (SLC25A24-WT) alongside mutant variants of these regions (SLC25A24-MUT) were synthesized (Shanghai Transheep Biotech Co., Ltd.). These constructs were co-transfected into KGN cells lines utilizing Lipofectamine™ 3000 Transfection Reagent (Invitrogen), with assays conducted 48 h post-transfection to assess luciferase activity via a dual-luciferase reporter gene system.
Cell viability assay
For the Cell Counting Kit-8 (CCK-8) assay, which is used to assess cell viability based on mitochondrial activity, 5,000 KGN cells per well were seeded in a 96-well plate and allowed to adhere for 24 h. Post-transfection with either siRNA targeting SLC25A24 or a control siRNA, cells were further incubated to allow for gene knockdown effects. Subsequently, 10 µL of CCK-8 solution (Sant Biotechnology, Shanghai, China) was added to each well, and the plate was incubated for 1 h at 37 °C. Absorbance was measured at 450 nm using an enzyme-linked immunosorbent assay (ELISA) reader to assess cell viability. All assays were conducted in triplicate to ensure consistency and reliability of the results.
TUNEL
To detect apoptotic cells in KGN cell samples, the TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) assay was performed. Cells were cultured on coverslips, fixed with 4% paraformaldehyde for 30 min, and permeabilized with 0.1% Triton X-100 on ice for 2 min. After incubation with the TUNEL reaction mixture at 37 °C for 60 min in the dark, cells were counterstained with DAPI to label nuclei. TUNEL-positive cells, emitting green fluorescence, were observed under a fluorescence microscope, and the percentage of apoptotic cells was calculated by comparing the number of TUNEL-positive cells to the total number of DAPI-stained cells. This assay provided insights into the effects of miR-361-5p and SLC25A24 on cell survival.
Statistical analysis
Data were expressed as mean ± standard deviation (SD). Comparative analyses among multiple groups employed ANOVA tests. SPSS software version 22.0 (IBM Corporation, USA) was utilized for all statistical computations, with a P-value of less than 0.05 denoting statistical significance.







留言 (0)