
記住我
Dear Editor,
Incontinentia pigmenti (IP), also known as Bloch-Sulzberger Syndrome (MIM # 308300), is an X-linked dominant genetic disease.1-3 The incidence of IP in newborns ranges from 1:50000 to 1:40000.3 This disease is more commonly seen in females, as it is usually lethal in males.
IP is caused by a loss of function variants in the inhibitor of nuclear factor kappa B kinase subunit gamma (IKBKG) gene. About 70–80% of IP patients may present with deletions of exon 4 to exon 10 in the IKBKG gene and 30% with microdeletions, missense, frameshift, nonsense, and splice site variants.4 Due to the presence of the highly homologous non-functional pseudogene IKBKGP1 (inhibitor of nuclear factor kappa B kinase subunit gamma pseudogene 1), next-generation sequencing cannot be reliably used for molecular diagnosis of IP.5 For suspected IP patients targeted mutation analysis of the IKBKG gene should be performed. Detection of deletions of exon 4 to exon10 in the IKBKG gene should be performed initially; later, if it is confirmed that the exon 4 to exon 10 deletion does not exist, Sanger sequencing can be considered for sequence analysis of the entire IKBKG gene.
Here, we design two novel detection methods for molecular diagnosis of IP [Supplementary Figure 1, Supplementary Materials and Methods].
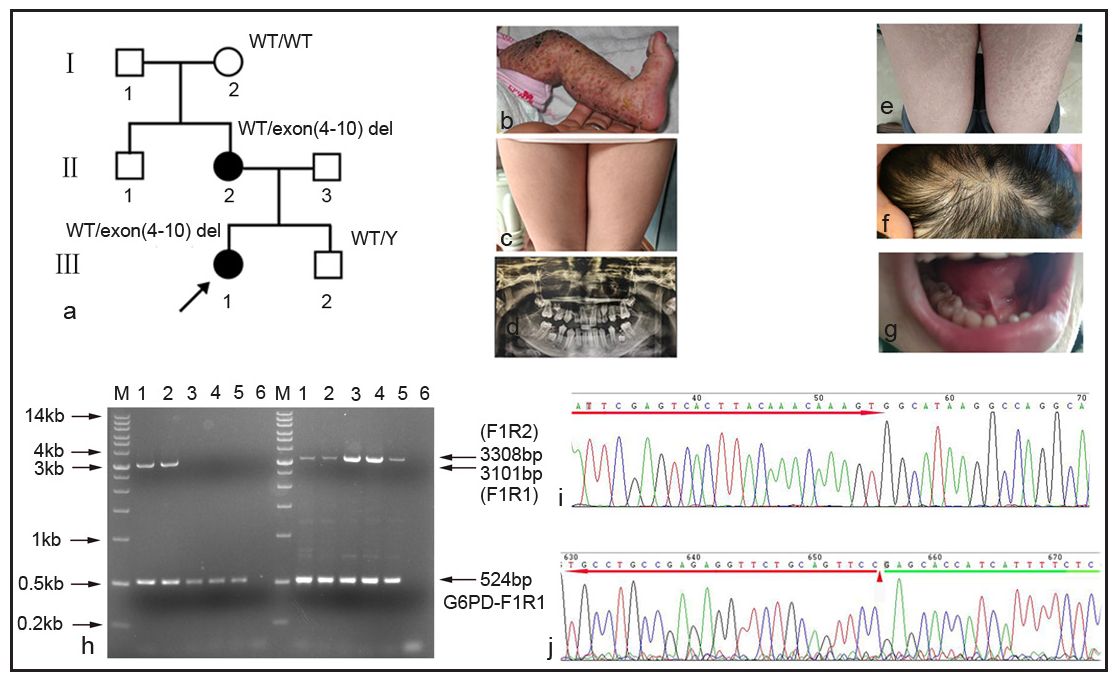
In family 1 [Figure 1a], the proband (Ⅲ1, 3 months, female) exhibited many clinical symptoms: blisters and ulceration on the left leg from birth [Figure 1b], textured stripes on the skin [Figure 1c], loss of teeth [Figure 1d], pigmentation of the entire skin and sparse scalp hair. The proband’s mother also presented with similar clinical symptoms: pigmentation of the entire skin [Figure 1e], sparse hair on the scalp [Figure 1f], loss of teeth [Figure 1g], and thinning of the nail plate.
Export to PPT
The proband’s mother was pregnant again, and hence a prenatal diagnosis with amniotic fluid was done. Subsequently, a gap-PCR (Polymerase chain reaction) was carried out to identify the large-scale (approximately 11.7 kb) exon 4 to exon 10 deletion in the IKBKG (NM_001099857.5) gene. The agarose electrophoresis results showed that both the proband and the proband’s mother carried one wild-type allele (3308bp) and another exon 4 to exon10 deletion allele (3101bp). Meanwhile, only wild-type alleles (3308bp) were observed in the grandmother, amniotic fluid, and normal male control. In addition, a 524bp amplicon was amplified with G6PD-F1R1 primer for internal control in all the sample lanes [Figure 1h]. Further, the exon 4 to exon10 deletion amplicon (3101bp) was purified for Sanger sequencing to confirm the specificity of the amplification. The amplicon was specific for IKBKG by sanger sequencing [Figure 1i]. Because there were two identical MER67B repeat sequences in exon 3 and downstream of exon 10, it was unable to identify specific breakpoints by Sanger sequencing [Figure 1j]. The proband’s mother had a healthy male child.
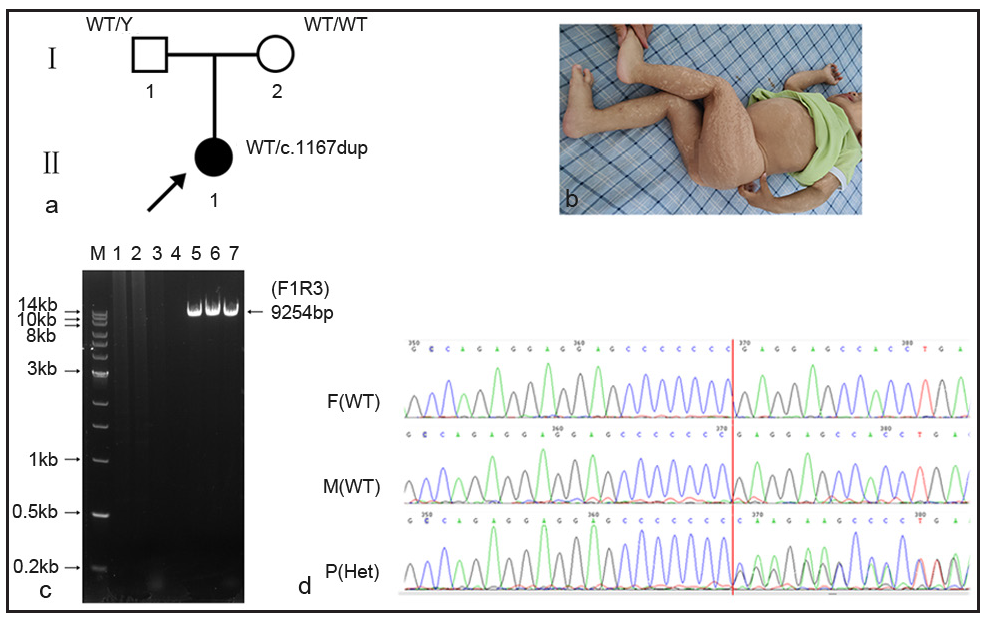
In family 2 [Figure 2a], clinical features of the proband (Ⅱ1, 1- year old, female) revealed generalised pigmentation [Figure 2b], short stature, growth delay, motor development delay, and enlarged lymph nodes in the left armpit and occipital region. Other laboratory investigations included white blood cells: 22.8×109/L (5.0–14.2×109/L), neutrophils: 11.76×109/L (0.8–6.1×109/L), monocytes: 2.61×109/L (0.2–1.14×109/L), immunoglobulin E: 1040 IU/ml (1–100 IU/ml), immunoglobulin G: 18.2 g/l (7–16 g/l) and immunoglobulin M: 3.54 g/l (0.4–2.3 g/l).
Export to PPT
Whole exome sequencing (WES) showed a pathogenic variant in the IKBKG gene, chrX:153792576-153792577 (GRCh37), NM:001099857.5: exon10, c.1167dupC (p.Glu390fs), which was reported in the ClinVar database (Variation ID: 372387). Due to the presence of a highly homologous pseudogene IKBKGP1 in the genome, long and accurate polymerase chain reaction (LA-PCR) was used to verify the authenticity of the variant.
LA-PCR results showed that the expected amplicon (9254bp) was observed in all the sample lanes [Figure 2c]. The three amplicons were purified for Sanger sequencing using sequencing primers (seq_exon10-R). Variant c.1167dupC was found in the proband, but was absent in parents [Figure 2d].
Our primer design is different from the previous published literature.6 our method can simultaneously amplify with two pairs of primers, one of which is an internal control primer, ensuring the success of PCR reaction and the other can specifically amplify the target region. In addition, our novel detection method has significant economic benefits for the diagnosis of IP patients, especially for prenatal diagnosis.
留言 (0)