
記住我
Streptococcus pneumoniae is a common opportunistic pathogen that asymptomatically colonizes the human upper respiratory tract. However, this process may result in severe invasive diseases including pneumonia, meningitis, and bacteremia in immunocompromised or environmentally stressed patients (Bogaert et al., 2004). S. pneumoniae pathogenicity stems from its ability to evade immune system clearance and adapt within the host thus contributing to its status as a major global public health concern (O’Brien et al., 2009; Prina et al., 2015; Andre et al., 2017). This pathogenicity of S. pneumoniae is also contingent upon colonization and virulence factors including capsular polysaccharides (CPS), surface proteins, and proteases (Kadioglu et al., 2008; Mitchell and Mitchell, 2010). In particular, the CPS is a pivotal virulence determinant due to its dual role in protection from host immune defenses and in promoting efficient colonization and invasion of host tissues (Kadioglu et al., 2008; Geno et al., 2015). Regulation of CPS synthesis includes transcriptional regulation of cps promoters (Shainheit et al., 2014; Wen et al., 2015; Wen et al., 2016), epigenetic regulation of phase variation (Wang et al., 2020; Zhang et al., 2021), and post-translational modification of the bacterial tyrosine kinase system CpsBCD (Morona et al., 2000; Nourikyan et al., 2015; Nakamoto et al., 2021).
Transcriptional regulation of capsular expression plays a crucial role in the pathogenesis of pneumococcal disease and is a key pathway that modulates CPS production (Shainheit et al., 2014; Wen et al., 2015; Wu et al., 2016). The core −10 and −35 motifs and the spacing between these elements are key regulators of the transcriptional response (Shainheit et al., 2014; Wen et al., 2015; Wen et al., 2016). The involvement of these promoter elements has been illustrated for the serotype 2 strain D39 (Wen et al., 2015) and the serotype 4 strain TIGR4 (Shainheit et al., 2014). Additionally, the absence of the core promoter elements results in a marked reduction in CPS production and a concomitant decline in virulence in murine experimental infection models (Wen et al., 2015).
Regulatory factors that directly interact at the cps promoter sequence are only partially defined (Moscoso and García, 2009) but include ComE (Zheng et al., 2017), ComX (Luo and Morrison, 2003), MgaSpn (Xiao et al., 2021), RegM (Giammarinaro and Paton, 2002), and CpsR (Wu et al., 2016). These regulators perform differing functions but all are necessary for the full extent of capsule synthesis. For instance, the phosphorylated form of ComE negatively regulates cps gene cluster expression in vivo (Zheng et al., 2017). The MgaSpn protein in S. pneumoniae is orthologous to the Mga/AtxA family member Mga found in Streptococcus pyogenes and functions as an environmental sensor to regulate capsule and teichoic acid production (Xiao et al., 2021). In addition, cps transcriptional regulators from serotype 3 S. pneumoniae strains can function in non-serotype 3 strains indicating a shared regulatory mechanism across strains (Moscoso and García, 2009).
Our laboratory had previously identified a novel CPS regulatory factor SPD_0410 in serotype 2 strain D39 (Wu et al., 2016). In the current study, we further characterized this protein and found it functioned as a negative regulator of CPS production. This protein directly bound the cps promoter to modulate CPS expression and influenced bacterial virulence. We also linked SPD_0410 to regulation of glycolysis and gluconeogenesis and the presence of glucose eliminated the regulatory ability of SPD_0410. This study links SPD_0410 with the CPS regulatory network and highlights its role in virulence and provides novel perspectives and insights into the exploration of hypothetical proteins associated with S. pneumoniae virulence.
2 Materials and methods 2.1 Bacterial strains and growth conditionsThe bacterial strains and plasmids used in this study are listed in Table 1. S. pneumoniae strain D39 derivatives were cultured in C+Y medium (C medium supplemented with 5% yeast extract pH 7.0) in the presence or absence of glucose as sole carbon source as required or on blood agar plates at 37°C in a 5% CO2 atmosphere. Antibiotics were used at the following concentrations: erythromycin 100 μg/mL. streptomycin 150 μg/mL, kanamycin 200 μg/mL as well as spectinomycin 50 μg/mL for Escherichia coli and 200 μg/mL for S. pneumoniae.
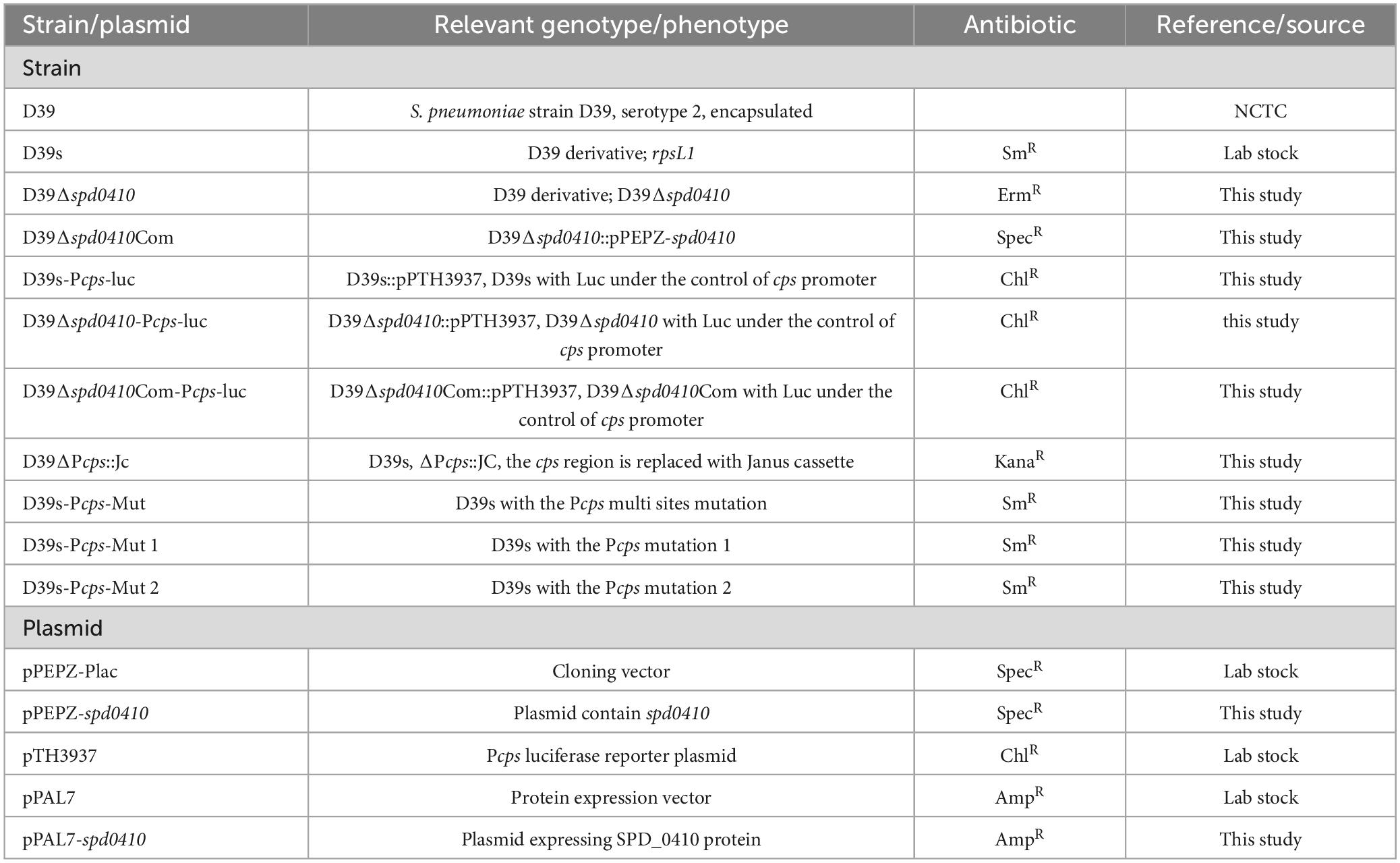
Table 1. Strains and plasmids.
2.2 Construction of mutantsMutants were generated via homologous recombination using standard techniques using the primers specified in Supplementary Table 1. All strains employed in this study were derived from D39s, a streptomycin-resistant S. pneumoniae serotype 2 variant (Zhang et al., 2021). Construction of mutant strain D39Δspd0410 utilized D39s genomic DNA as a template. In brief, primers P1/P2 and P3/P4 were used to amplify the upstream and downstream fragments of the spd0410 gene by PCR and Erm F/Erm R primers to amplify the erm fragment with homologous arms at both ends. These three amplicons were ligated to generate a recombinant fragment that was then introduced into D39s by transformation. Individual colonies from blood agar plates containing erythromycin were isolated and mutants were screened via PCR using primers P1/P4 to identify erythromycin-resistant D39Δspd0410 mutants.
The plasmid pPEPZ and the entire spd0410 gene fragment from above were double digested with BgI II and XhoI and ligated to produce pPEPZ–spd0410. This plasmid construct was then transformed into D39Δspd0410 to obtain the ectopic complemented strain D39Δspd0410Com. The spd0410 gene was integrated into the bacterial genome via plasmid pPEPZ, ensuring its proper expression and functionality within the genome.
To construct cps promoter (Pcps) point mutants, we amplified the Janus cassette fragment (Sung et al., 2001) with primers Pr1332/Pr1333 as well as upstream and downstream regions of cps promoter fragments with primers Pr9020/9026 and Pr9027/Pr9023. These three amplicons were ligated and transformed into D39s to generate D39ΔPcps::JC. Subsequently, we used D39s as a template to amplify the upstream and downstream regions of cps promoter fragments with Pr9028/Pr9021 and Pr9022/Pr9029. We connected the two fragments to generate a Pcps fragment of 7-point mutants. The fragment was then transferred to D39ΔPcps::JC to produce the D39Pcps-mut 1 strain. As mentioned above, primers Pr9028/Pr9025 and Pr9024/Pr9029 were used to amplify the upstream and downstream fragments of cps promoter fragments containing 8-point mutations, and then the fragment was connected and transferred into D39ΔPcps::JC to produce D39Pcps-mut 2 strain. D39Pcps-mut 1 and D39Pcps-mut 2 were used as templates to amplify upstream and downstream regions of the cps promoter using the primers Pr9028/Pr9034 and Pr9033/Pr9029, respectively. The two fragments were then ligated and transferred into the D39ΔPcps::JC strain resulting in the generation of the binding site double mutant strain D39Pcps mut.
2.3 Evolutionary analysisThe protein BLAST tool used the amino acid sequence of SPD_0410 from S. pneumoniae D39 to search the NCBI database. Multiple sequence alignment and cluster analysis of the top-scoring proteins were conducted using Clustal-X 2.1. The evolutionary tree of the proteins was constructed using MEGA 11. Phylogenetic analysis was performed using the Neighbor-Joining method, which applied the Poisson model to calculate pairwise distances. Bootstrap support values (%) were calculated with 1000 replicates to assess the reliability of the tree.
2.4 Western blottingProtein production was quantified from S. pneumoniae strains cultured in C+Y medium at 37°C / 5% CO2. The strains were harvested at logarithmic growth (OD600nm = 0.5) and lysed using a 0.5% deoxycholate buffer. The samples were separated using Tris-glycine SDS-PAGE and wet transferred to PVDF membranes. The membrane was incubated with 5% skim milk powder at 37°C for 2 h, then washed three times with TBST (Tris-buffered saline with 1% Tween 20) for 15 min each. After washing, the membrane was incubated overnight at 4°C with the primary antibody targeting CPS type 2 (1:500) (Danish National Serum Institute, Copenhagen). The CPS type 2 antibody was a non-adsorbed polyclonal antibody against type 2 polysaccharides, and the excess non-specific components in cell wall components were removed by the method described earlier (Kant et al., 2023). Following the overnight incubation, the membrane was washed again three times with TBST and then incubated with the secondary antibody, either goat anti-rabbit IgG or goat anti-mouse IgA (both at 1:10,000 dilutions), at 37°C for 1 h. After washing the membrane again, the gel image was performed. Protein bands were visualized using Image Lab software (Bio-Rad, Hercules, CA, USA) after incubation with Immobilon Western horseradish peroxidase (HRP) substrate peroxide solution (Millipore, Burlington, MA, USA).
2.5 Enzyme-linked immunosorbent assay (ELISA)Bacterial cells were collected during the logarithmic growth phase (OD600nm = 0.5) and suspended in a sterile PBS. These residues were then inactivated in a 58°C water bath for 45 min to prepare cell wall samples. After gradient dilution, the samples were coated onto 96-well plates and incubated overnight at 4°C. The primary antibody used was CPS polyclonal antibody type 2 (1:500) (see above) followed by incubation with goat anti-rabbit IgG-HRP antibody (1:500) (KPL, Gaithersburg, MD, USA). Absorbance at 450 nm was measured after color development to quantify CPS content on the bacterial surface. The results of the representative experiment were presented as mean ± standard deviation of three replicates.
2.6 Determination of capsular glucuronic acidLogarithmic growth phase samples were collected and washed twice with sterile PBS followed by an additional wash with Tris-MgSO4 to prepare the whole bacterial sample. Cell wall samples were prepared from bacterial precipitates, washed with sterile PBS, and then suspended in PBS containing pre-boiled 2% SDS and boiled at 100°C for 30 min. After cooling to room temperature, the suspension was centrifuged and the cell walls were washed 3 × with PBS to completely remove SDS. The resulting precipitate was suspended in 400 μL Tris-MgSO4 to which 40 μL TE buffer containing 30 mg/mL lysozyme was added for cell wall lysis. The quantification of glucuronic acid in cell walls or whole bacterial capsules was performed as previously described for the quantitative determination of uronic acid (Blumenkrantz and Asboe-Hansen, 1973).
2.7 Transmission electron microscopy (TEM)Bacterial precipitates harvested during logarithmic growth in C+Y medium were washed twice with sterile PBS. Then the residues were gently resuspended in 1 mL 2.5% glutaraldehyde fixing solution added slowly along the tube wall to ensure minimal disruption to the bacteria and then fixed overnight at 4°C. Electron microscopy was performed by Xavier Biotechnology (Wuhan, China). Capsule thickness was determined by measuring 15 randomly chosen cells using Image J software.
2.8 Expression and purification of SPD_0410 proteinThe S. pneumoniae spd0410 gene was amplified by PCR and cloned into expression vector pPAL7 using EcoRI and XhoI double digests of amplicons (see above). The construct was transferred into E. coli BL21(DE3) for fusion protein expression. SPD_0410 recombinant protein was induced in a Luria-Bertani medium supplemented with 100 μg/mL ampicillin and 0.5 mM isopropyl-b-d-thiogalactoside (IPTG). The protein was purified using the commercial Profinity eXact system (Biorad, Hercules, CA, USA).
2.9 Electrophoretic mobility shift assay (EMSA)Primers PcpsF/PcpsR were employed to amplify fragments of the cps promoter region from strain D39s (Supplementary Table 1). The binding experiment was initiated by incubating a 10 μL reaction mixture at 25°C for 10 min comprised of 1 μL binding buffer (10 mM Tris-HCl, pH 7.4, 1 mM dithiothreitol, 1 mM EDTA, 50 mM KCl, 5% glycerol, 50 mg/mL bovine serum albumin, 0.05% Nonidet P-40), 0.5 μg of poly(dI-dC), 0–6 μg of SPD_0410 protein, and 1 ng of labeled probe. Following this incubation, a 100-fold excess of unlabeled probes was added to serve as specific competitors in the cold probe reaction system. The mixture was then further incubated at 25°C for 20 min followed by electrophoresis through 5% native polyacrylamide gels in 1 × TBE buffer. Subsequent steps followed the protocol provided by the LightShift R Chemiluminescence EMSA Kit (Thermo Fisher, Pittsburg, PA USA).
2.10 DNase I footprintingDNase I footprinting assays were performed as described previously (Wang et al., 2012). The resulting probe (300 ng) was briefly incubated with varying amounts of protein in a 40 μL reaction system at 25°C for 30 min. Following incubation, the reaction samples underwent enzyme digestion using 0.015 units of DNase I (Promega. Madison, WI, USA) at 25°C for 1 min. After enzyme digestion, the proteins were removed via phenol extraction, and the DNA was precipitated with ethanol. The precipitated DNA was dissolved in Milli-Q ultra-pure water and analyzed using an ABI sequencer and ABI GeneScan 500 Liz was used as the ladder.
2.11 RNA extraction and qRT-PCRTotal RNA was extracted from S. pneumoniae cultures harvested during the logarithmic (OD600nm = 0.5) or the early growth (OD600nm = 0.1) phases. Bacterial precipitates were suspended with 100 μL TE buffer containing 30 mg/mL lysozyme and incubated at 25°C for 25 min. Bacterial total RNA was subsequently extracted using a total RNA extraction kit designed for cultured cells/bacteria (Tiangen, Beijing, China). RNA (1 μg) was reverse transcribed into cDNA using the PrimeScript First strand cDNA synthesis kit (Takara Bio, Shiga, Japan). Real-time PCR was conducted on a CFX Connect system (Biorad) using the gyrB gene as the internal reference standard. All the primers used in the experiment are shown in Supplementary Table 1. The results of representative experiments were expressed as the mean ± standard deviation of three replicates.
2.12 Construction of luciferase reporter strains and luciferase assayThe integrating plasmid pTH3937 (Wen et al., 2015) containing the complete promoter region and firefly luciferase gene was transferred into D39s, D39Δspd0410, and D39Δspd0410Com strains using bacteria grown to OD600nm = 0.1 in C+Y medium at 37°C. The plasmid was supplied by Professor Jingren Zhang from Tsinghua University, Beijing. The fluorescence intensity (RLU) was measured by mixing 100 μL of bacterial culture with 0.66 μM fluorescein sodium salt. The results were normalized to the optical density (OD600nm) of the samples.
2.13 RNAseq analysisS. pneumoniae strains D39s and D39Δspd0410 strains were cultured in C+Y medium to OD600nm = 0.5. Bacterial total RNA was extracted using the RNAprep pure cell/bacterial kit (Tiangen, Beijing, China) followed by transcriptome sequencing conducted at Novogene Bioinformatics Technology (Beijing, China). The reference genome used was the S. pneumoniae D39s genome (GenBank Acc. No. NC_008533.2). Differential expression analysis of two groups (each with 3 biological replicates) was performed using the DESeq2 R package (version 1.20.0). Genes with adjusted Padj < 0.05 identified by DESeq were classified as differentially expressed.
2.14 Adhesion and antiphagocytic assayAdhesion and invasion assays of S. pneumoniae were conducted using the human type II lung epithelial cell line A549. Cells were seeded at a density of 2 × 105 per well in 24-well plates and cultured overnight at 37°C. The confluent epithelial cell monolayers (2 × 105 cells/well) were then inoculated with 2 × 107 colony-forming units (CFU) of pneumococci at a multiplicity of infection (MOI) of 1:100. The cultures were incubated in Dulbecco’s Modified Eagle’s medium (DMEM) at 37°C with 5% CO2 for 1 h. After incubation, the cells were washed 5 × with sterile PBS and lysed by the addition of double-distilled water for the adhesion experiment. The resulting lysate was diluted and plated onto agar plates to enumerate bacteria inside and outside the cells. Invading bacteria were enumerated by exposing extracellular bacteria to 100 μg/mL penicillin and 10 g/mL gentamicin for 15 min, respectively. The number of intracellular bacteria was then determined using the abovementioned method.
Primary peritoneal macrophages from male C57BL/6 mice were extracted and isolated according to the method previously described (Zhong et al., 2019). After 4–5 days of paraffin injection into the peritoneal cavity of mice, macrophages were extracted from the peritoneal lavage and inoculated into 24-well plates at a density of 2 × 105 cells/well. Phagocytosis was evaluated using primary peritoneal macrophages from a mic, employing a protocol identical to that of the adhesion experiment. The anti-phagocytosis rate was calculated using the following formulas:
Anti-phagocytosisrate(%)
=extracellularCFUintracellularCFU+extracellularCFU
2.15 Animal experimentsMale C57BL/6 mice (6–8 weeks old, weight 18–20 g) were procured from the Laboratory Animal Center of Chongqing Medical University. All animal experiments described in this study were approved by the Animal Care and Use Committee of Chongqing Medical University and were conducted strictly with the provisions of the Guidelines for the Care and Use of Experimental Animals.
To investigate the effects of SPD_0410 on the colonization of the mouse nasopharynx, mice were anesthetized by peritoneal injection of 2% pentobarbital sodium (50 mg/kg), and the bacteria were inoculated through the nasal cavity to establish a pneumonia model. Mice were randomly divided into two groups (n = 6) and each mouse was inoculated nasally with 2 × 107 CFU/30 μL bacteria. Nasal lavage fluid, heart blood, spleen, and lung tissues were collected 48 h after infection. After proper dilution, the samples were placed on blood agar plates to determine CFU.
The role of SPD_0410 in systemic infection was explored by dividing the mice into two groups (n = 12). Each mouse was intranasally infected with 1 × 108 CFU/30 μL of bacteria. Survival was monitored daily for 14 days. After the 14-day survival observation period, the surviving mice were anesthetized by intraperitoneal injection of 2% pentobarbital sodium (50 mg/kg) and euthanized via cervical dislocation.
Lung biopsy samples were collected from mice infected with sterile PBS, D39s, and D39Δspd0410 strains, sterile PBS simulated infection as a control. Lung tissues were initially fixed in 4% paraformaldehyde at 4°C for 48 h followed by embedding in paraffin and sectioning into 5 mm thick slices. Sections were subsequently stained with hematoxylin and eosin (H&E) using standard protocols.
2.16 Statistical analysisAll statistical analyses in this study were conducted using Prism 8 software (GraphPad Software, San Diego, CA, USA). The data were evaluated using either an unpaired two-tailed Student’s t-test or a non-parametric Mann–Whitney U test. Survival data were analyzed using the log-rank (Mantel-Cox) test. Statistical significance was defined as ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant.
3 Results 3.1 Multi amino acid sequence alignment and evolutionary tree analysisSPD_0410 was previously identified as a potential transcriptional regulator that influenced CPS regulation in S. pneumoniae strain D39 (Wu et al., 2016). The protein is highly conserved among Gram-positive bacteria, particularly within the Streptococcus genus (Figure 1). However, these orthologs were primarily classified as hypothetical and their specific biological functions have yet to be thoroughly characterized. The data also revealed that the orthologous proteins SPD_0410, WP_035342599, and WP_077140481 possessed helix-turn-helix DNA binding domains (Carranza et al., 2024) suggesting that SPD_0410 may also have a function related to DNA binding.
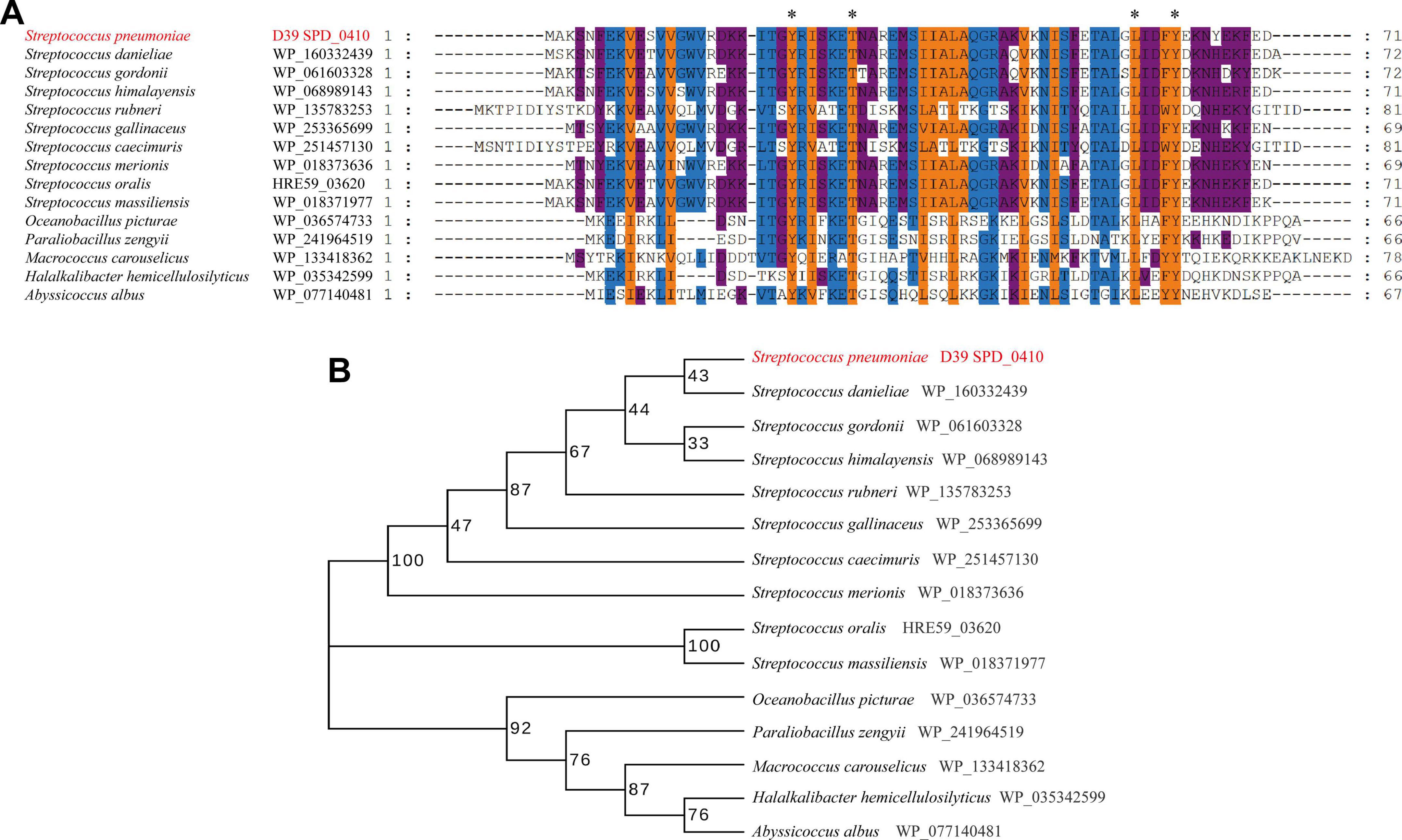
Figure 1. Conserved and evolutionary analysis of SPD_0410. (A) Multiple sequence alignment of SPD_0410 with homologous proteins in various bacterial species. The residues labeled with * represent the invariant residues. (B) Evolutionary tree of SPD_0410 protein and its homologous proteins.
3.2 SPD_0410 affects CPS production by down-regulation of the cps operon transcriptionTo investigate the role of SPD_0410 in CPS expression, we constructed the spd0410 defective (D39Δspd0410) and complemented (D39Δspd0410Com) strains (Supplementary Figures 1A, B). The growth rates in C+Y medium for these strains did not significantly differ (Supplementary Figure 1C). We subsequently assessed the relative CPS levels in these strains and CPS levels in strain D39Δspd0410 were significantly elevated compared to the WT strain D39s. WT levels were also restored in the complemented strain D39Δspd0410Com (Figure 2A). These differences were further verified using ELISA assays that quantified surface CPS levels (Figure 2B). The S. pneumoniae serotype 2 capsule comprises uronic acid (Kolkman et al., 1998) so we also quantified glucuronic acid levels in these strains. The mutant strain D39Δspd0410 produced whole cell and cell wall-associated uric acid levels significantly greater than the WT or complemented strains (Figures 2C, D). We also measured capsule thickness in these test strains using 15 capsules per group measured randomly. We found a significant increase in capsule thickness in the D39Δspd0410 strain (57.6 ± 8.4 nm) versus D39s (47.5 ± 10.4 nm) and D39Δspd0410Com (50.3 ± 9.4 nm) (Figure 2E). These findings strongly indicated that deletion of the spd0410 gene markedly enhanced CPS production and its presence at the bacterial cell surface.
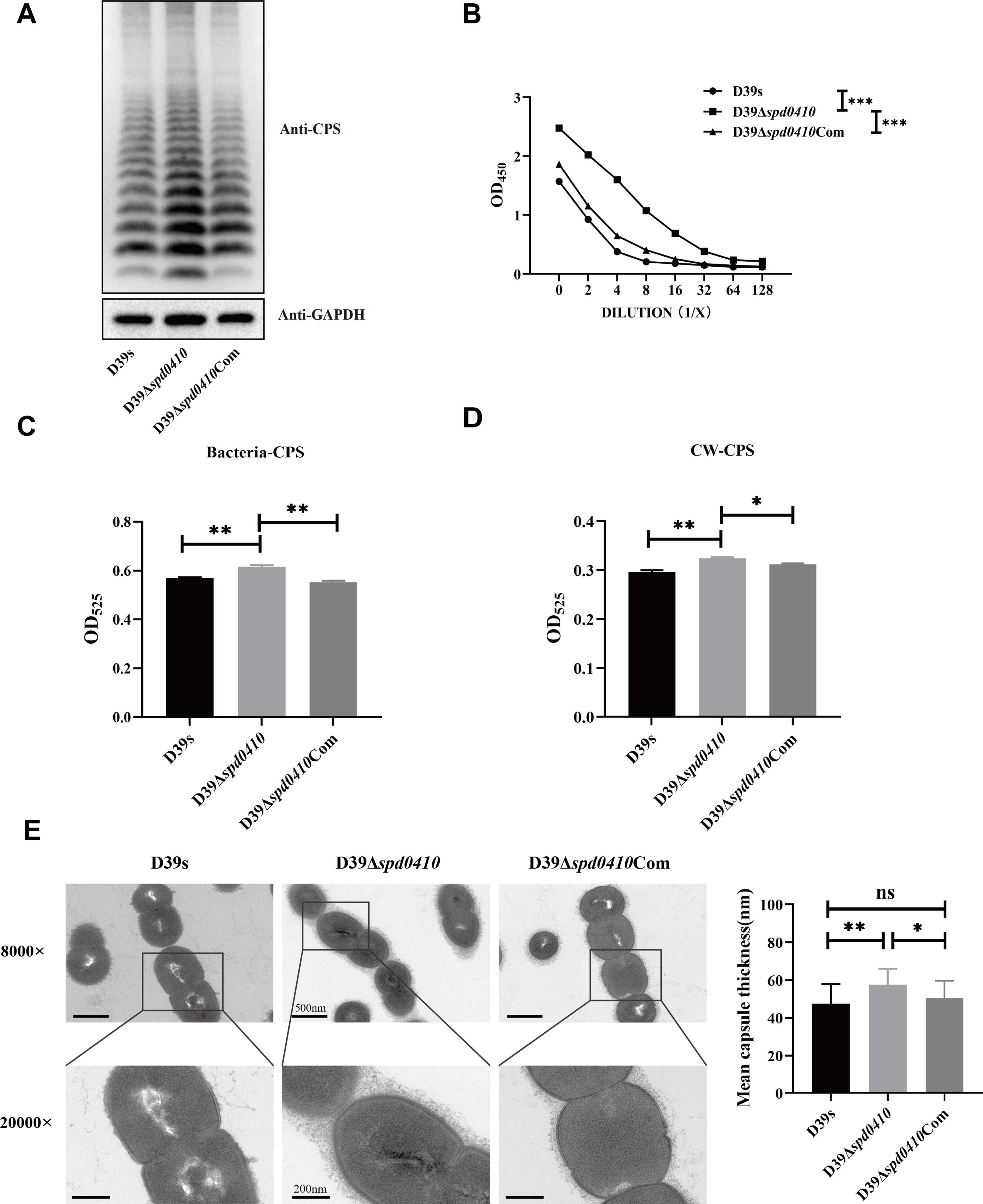
Figure 2. Effect of deletion of spd0410 gene on CPS synthesis. (A) Western blotting detected CPS expression in the indicated strains’ whole-cell lysates. GADPH was used as a sample loading control. (B) ELISA determined the content of the capsule in bacterial lysate. Western blots and ELISA were probed with a rabbit anti-serotype 2 CPS polyclonal antibody. (C,D) Determination of capsule content by uronic acid assay. (C) Comparisons of whole-cell CPS levels. (D) Comparisons of cell wall-associated CPS levels. (E) Transmission electron microscopy of strains. The mean capsule layer diameters are indicated; n = 15. The average capsule thickness of D39s, D39Δspd0410 and D39Δspd0410Com is 47.5 ± 10.4, 57.6 ± 8.4 and 50.3 ± 9.4 nm, respectively; The results of representative experiments are presented as the mean of three replicates ± SD. ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant, as analyzed by unpaired two-tailed Student’s t-test.
We further examined the regulation of the cps operon by monitoring steady-state mRNA expression of the cps2A-D genes to ascertain whether SPD_0410 regulates the expression of the cps operon. We found no significant differences in cps operon expression between D39s and D39Δspd0410 during logarithmic growth (OD600nm = 0.5) (Supplementary Figure 2). In contrast, the D39Δspd0410 strain displayed higher levels of mRNA for cps2A-D genes versus D39s at early growth stages (OD600nm = 0.1) (Figure 3A). Together, these results indicated that SPD_0410 is a negative regulator that is may also linked to metabolic and resource allocation mechanisms of bacterial growth. Deletion of spd0410 could probably activate specific transcriptional regulatory mechanisms in the early growth phase to facilitate environmental adaptation, altering the cps gene cluster levels and making new adjustments during logarithmic growth.
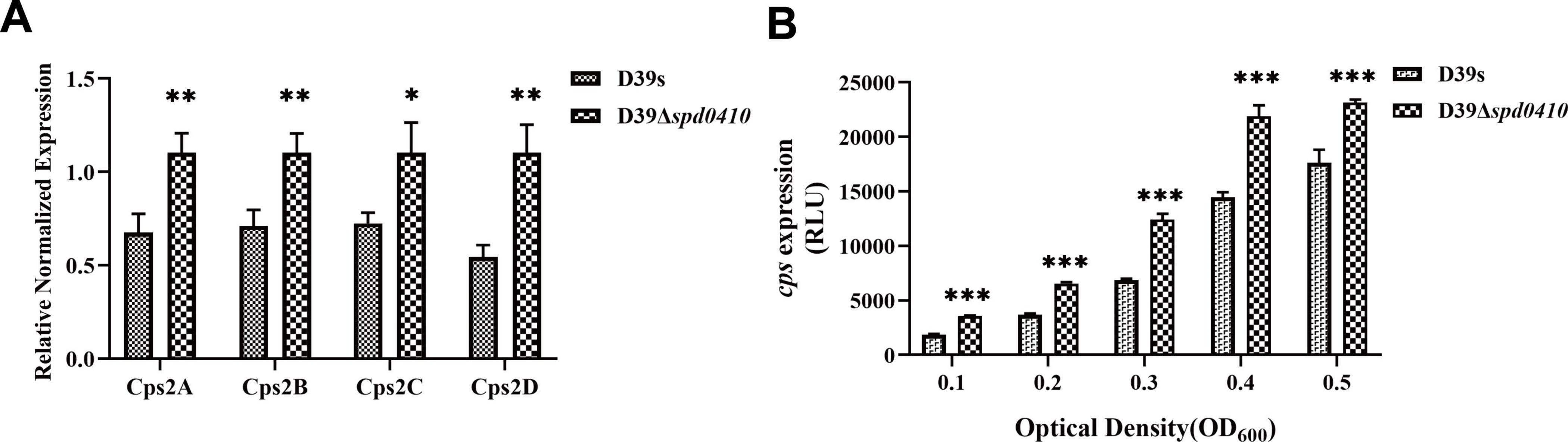
Figure 3. Transcriptional levels of cps gene clusters in D39s and D39Δspd0410 strains. (A) Relative mRNA levels of cps2A-D in early stages of bacterial logarithmic growth (OD600nm = 0.1), the first four genes downstream of the cps operon in the strains. Relative mRNA levels are expressed relative to that of gyrB. (B) Luciferase activity analysis evaluated the expression of cps promoters in D39s and D39Δspd0410 strains. The results of representative experiments are presented as the mean of three replicates ± SD. ***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant, as analyzed by unpaired two-tailed Student’s t-test.
To characterize the activity of the cps promoter region during growth, the firefly luciferase gene was fused with the cps promoter and introduced into the D39s and D39Δspd0410 strains. We found that cps promoter activity was significantly enhanced in D39Δspd0410 (Figure 3B). This result was not entirely consistent with the mRNA data likely because the luciferase reporter assay only reflected the activity of the cps promoter region and did not fully capture the actual decay or stabilization mechanisms of the mRNA itself. However, it was clear that SPD_0410 negatively regulates CPS biosynthesis.
3.3 SPD_0410 specifically binds to the cps promoter region and regulates CPS productionTo ascertain whether SPD_0410 specifically binds to the cps promoter region in vitro, we initially expressed and purified the SPD_0410 protein from S. pneumoniae using a heterologous expression system (Figure 4A). DNA electrophoretic mobility shift assays (EMSA) using 5′-biotin-labeled 218-bp long cps promoter probes indicated that electrophoretic mobility of the protein-DNA complexes decreased as the SPD_0410 protein concentration was increased (Figure 4B). Moreover, the introduction of unlabeled cold probes into the binding mixture led to reduced migration of the protein-DNA complexes (Figure 4B). These results indicated that SPD_0410 specifically binds to the cps promoter region. We additionally performed DNase I footprinting using the same 218-bp long cps promoter fragment labeled with FAM to determine the binding site more precisely (Figure 4C). We identified two protected sites on the cps promoter when SPD_0410 was present at 0.65 μg: a 24-bp region (5′-TCTGCTTCTAAAATATTGTTAGAA-3′) and a 25-bp region (5′-AAGATACTTAAAGATGCAGATAGTG-3′). These regions were upstream of the −35 and −10 boxes on the cps promoter (Figure 4D).
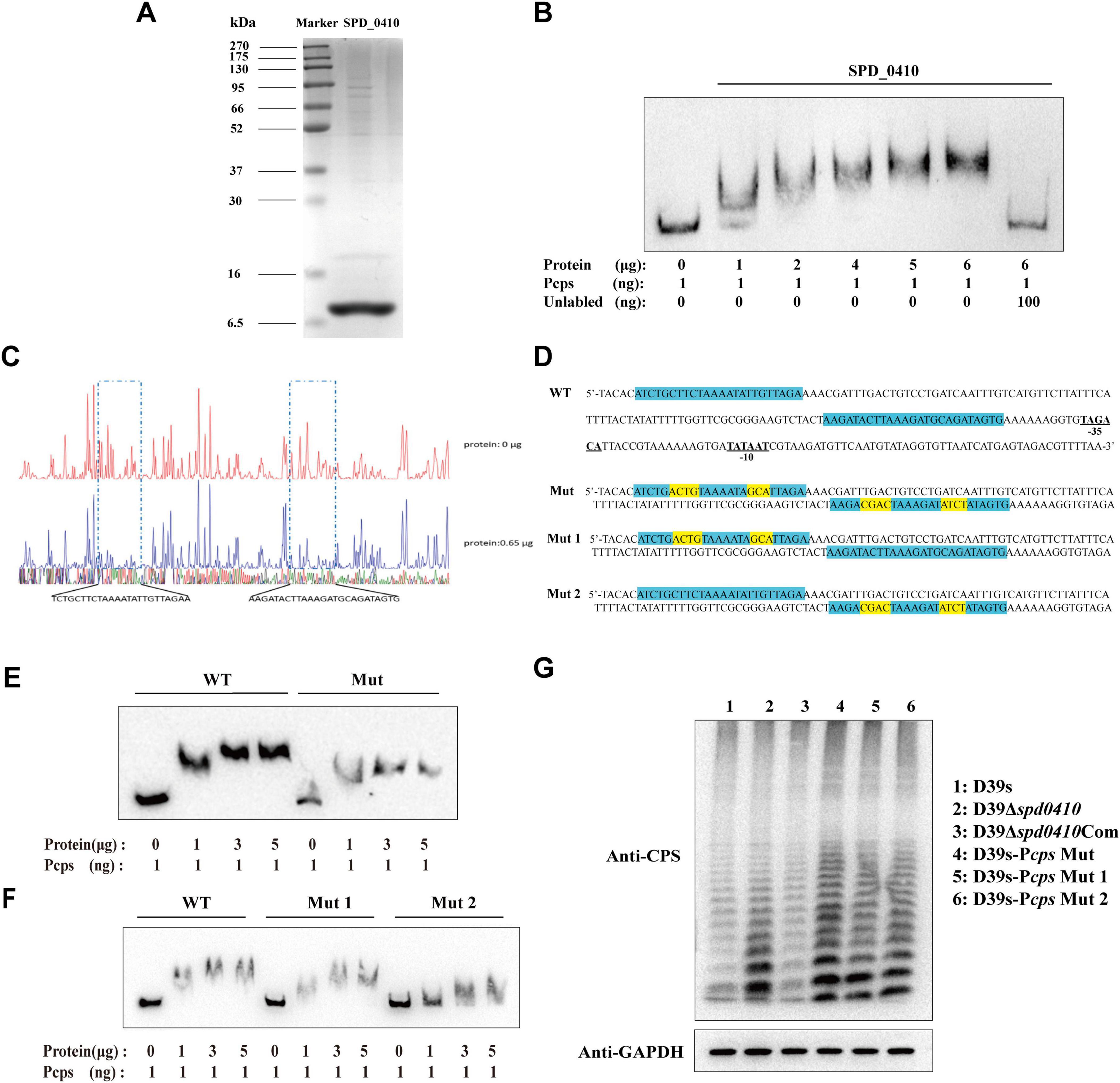
Figure 4. Electrophoretic mobility shift assay (EMSA) determines SPD_0410 binding to the Pcps. (A) SDS-PAGE analysis of the SPD_0410 protein. (B) EMSA of Pcps using the SPD_0410 protein, and 100 ng of non-biotin-labeled was added to compete with the labeled probe. (C) DNase I footprinting protection assay of SPD_0410. (D) Structural organization of the cps promoter-proximal region (WT). The binding sites for SPD_0410 are shown in blue. Mutated sequences are shown in yellow (Mut, Mut 1, and Mut 2). (E) Binding of SPD_0410 to the Pcps and Pcps Mut fragments. (F) Binding of SPD_0410 to the Pcps Mut 1 and Pcps Mut 2 fragments. (G) Western blotting was used to detect the expression of CPS in whole-cell lysates of the indicated strains.
To further confirm that SPD_0410 regulates CPS expression through specific binding to the cps promoter, we generated gene constructs containing 7 or 8 mutations at two distinct binding sites in vitro in the cps promoter sequences: Mut (the motif CTTCTAAAATATTG was mutated to ACGTTAAAATAGCA and the motif TACTTAAAGATGCAG was mutated to CGACTAAAGATATCT), Mut 1 (the motif CTTCTAAAATATTG was mutated to ACGTTAAAATAGCA) and Mut 2 (the motif TACTTAAAGATGCAG was mutated to CGACTAAAGATATCT) (Figure 4D). The binding affinity of SPD_0410 for the mutant Pcps probes was significantly reduced compared to the unmutated probe (Figures 4E, F). These data indicated that SPD_0410 specifically binds to the cps promoter.
Deletion or mutation of the −35 and −10 boxes within the core region of the cps promoter results in a substantial reduction in CPS production (Shainheit et al., 2014). We observed that SPD_0410 specifically binds to an area of the cps promoter located upstream of the −35 and −10 boxes (Figure 4D). We assumed the mutations we performed would not reduce CPS production. We introduced these point mutations into the bacterial genomes to test this hypothesis. We found that CPS was expressed at higher levels in D39s-Pcps-Mut, D39s-Pcps-Mut 1, and D39s-Pcps-Mut 2 compared to D39s (Figure 4G). This finding further reinforces the notion that SPD_0410 directly binds to the cps promoter and negatively regulates its expression, thereby impacting CPS synthesis. Additionally, these results complement previous studies and highlight the critical role of the cps promoter core region in regulating CPS production (Kadioglu et al., 2008).
3.4 SPD_0410 affects the glucose metabolism pathwayTo further elucidate the reasons behind the increased CPS levels resulting from the deletion of the spd0410 gene and to investigate its complete functionality, transcriptome sequencing was performed on both D39s and D39Δspd0410 strains. We found 79 transcripts that were significantly differentially expressed based on a cutoff value of 1.0-fold change and a Padj value of 0.05 in D39Δspd0410. In particular, 34 genes were increased and 45 genes were decreased (Figure 5A). The up-regulated genes were primarily associated with pyrimidine metabolism and virulence factors whereas the down-regulated genes were linked to the ABC transport system, the phosphotransferase system (PTS), and glucose metabolism (Table 2). For instance, enzyme II genes of the PTS responsible for mediating the internalization of extracellular carbohydrates (Marciniak et al., 2012) were down-regulated by 1.2-fold to 2.0-fold in D39Δspd0410 (Table 2). Glucose is the preferred carbon source for S. pneumoniae and is transported intracellularly by the mannose-type PTS (manLMN) (Carvalho et al., 2011; Bidossi et al., 2012). Our data indicated that the manLMN genes such as gadE, gadV, and gadW were significantly down-regulated by 1.7-fold to 1.9-fold in the D39Δspd0410 mutant (Table 2). These genes are also under carbon catabolic repression via CcpA, which inhibits the uptake and metabolism of non-preferred sugars by binding to catabolic response elements (cre) (Görke and Stülke, 2008; Carvalho et al., 2011). However, the levels of ccpA did not differ between the D39s and D39Δspd0410 (data not shown), suggesting that SPD_0410 and CcpA may interact synergistically in regulating carbon catabolism.
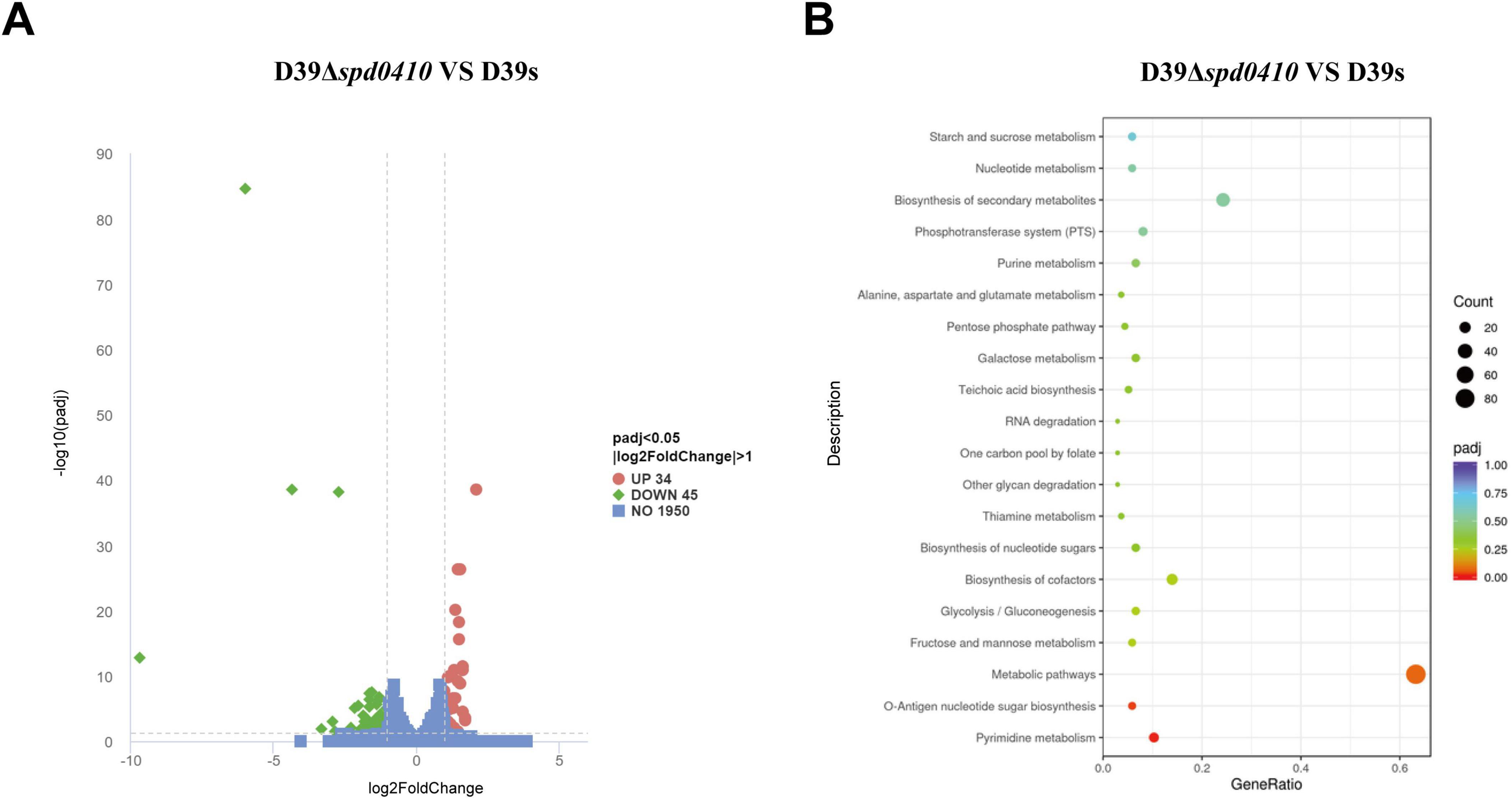
Figure 5. Comparison of differentially expressed genes between D39s and D39Δspd0410 by RNAseq analysis. (A) Volcano plot of differentially expressed genes. The abscissa represents the ratio of differentially expressed genes in D39Δspd0410 versus D39s; the ordinate represents the P-value between the two groups. (B) GO enrichment cluster analysis of differential genes.
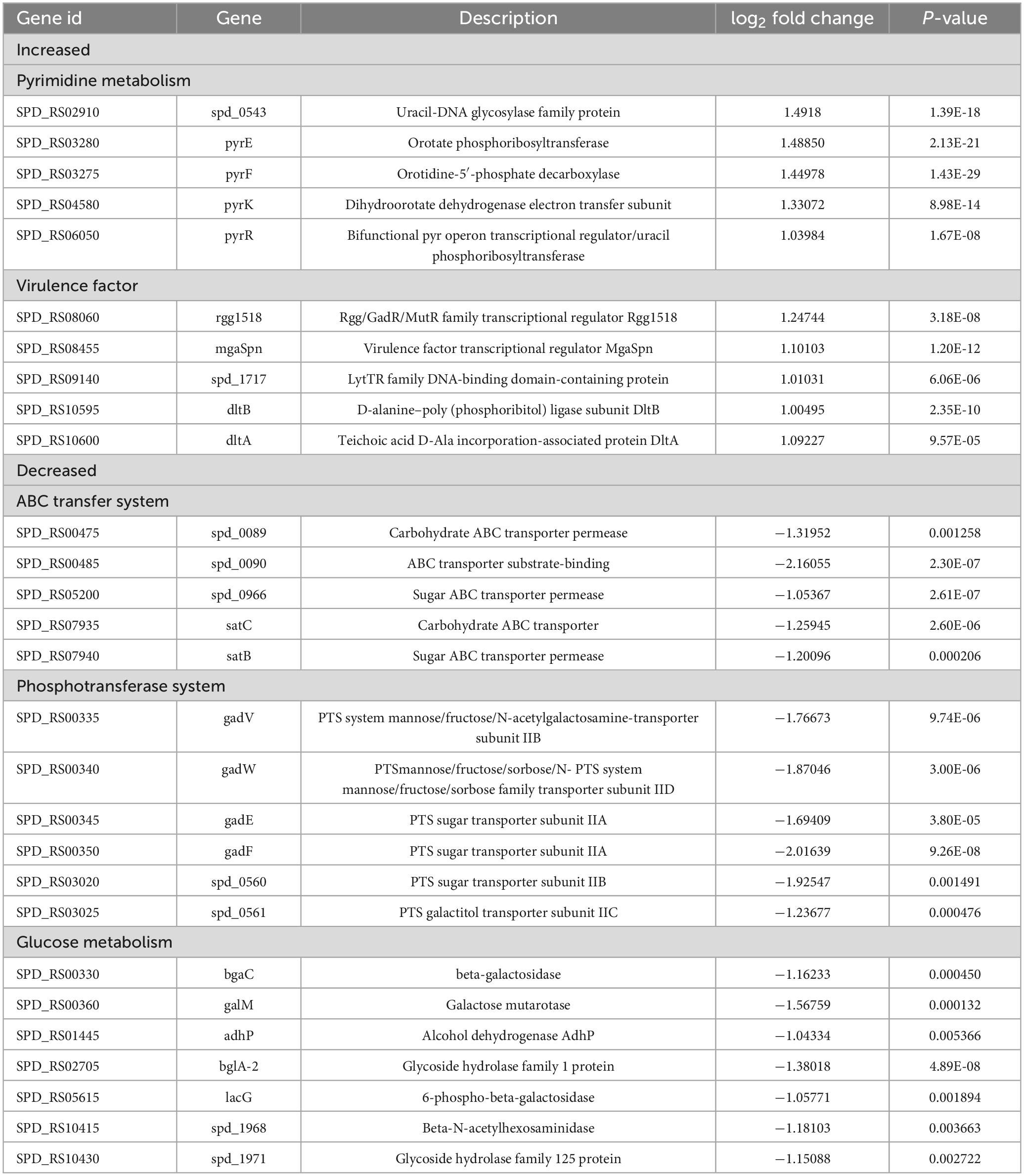
Table 2. Differential gene expression for D39Δspd0410 relative to the D39s by RNAseq analysis.
S. pneumoniae derives most of its ATP from glycolysis (Bättig and Mühlemann, 2008) where accumulated NADH is re-oxidized by NADH oxidase using oxygen and this process enhances glycolytic efficiency. Our data revealed changes in the expression of several glycolytic- and gluconeogenesis-related enzymes including galM, bglA-2, adhP, and spd1971 that were up-regulated by 1.0-fold to 1.6-fold (Figure 5B and Table 2). This indicated that intracellular energy metabolism is altered in D39Δspd0410 and links SPD_0410 to glucose transport and metabolism.
3.5 Glucose levels affect SPD_0410 regulation on CPSOur transcriptome results indicated that SPD_0410 can alter glucose metabolism (Figure 5B), so we examined whether glucose levels affected the growth of D39s and D39Δspd0410. We cultured strains in C+Y medium with glucose as the sole carbon source at levels of 4 mM, 8 mM, and 16 mM. We observed the growth of the strains at different glucose concentrations and found no significant differences between the two strains (Supplementary Figure 3). This indicated that D39Δspd0410 has strong adaptability, allowing it to rapidly sense changes in glucose levels in the environment and adjust cell metabolism accordingly. Consistent with these findings, bacterial morphologies were similar for both strains at all glucose levels, including in the absence of glucose (Supplementary Figure 4).
We further examined whether glucose might influence the regulation of CPS by SPD_0410. CPS levels were elevated as glucose levels were increased for the D39s strain but not the D39Δspd0410 strain (Figure 6A). The activity of the cps promoter in D39s-Pcps was increased as the glucose levels were increased. Still, this pattern was not observed in D39Δspd0410-Pcps (Figure 6B). In addition, we evaluated the CPS levels in the Pcps multi-site mutation strain D39s-Pcps-Mut and the results mirrored those of the mutant D39Δspd0410 (Figures 6C, D). These findings indicated that glucose is a regulator of SPD_0410. However, the specific mechanism needs to be further investigated.
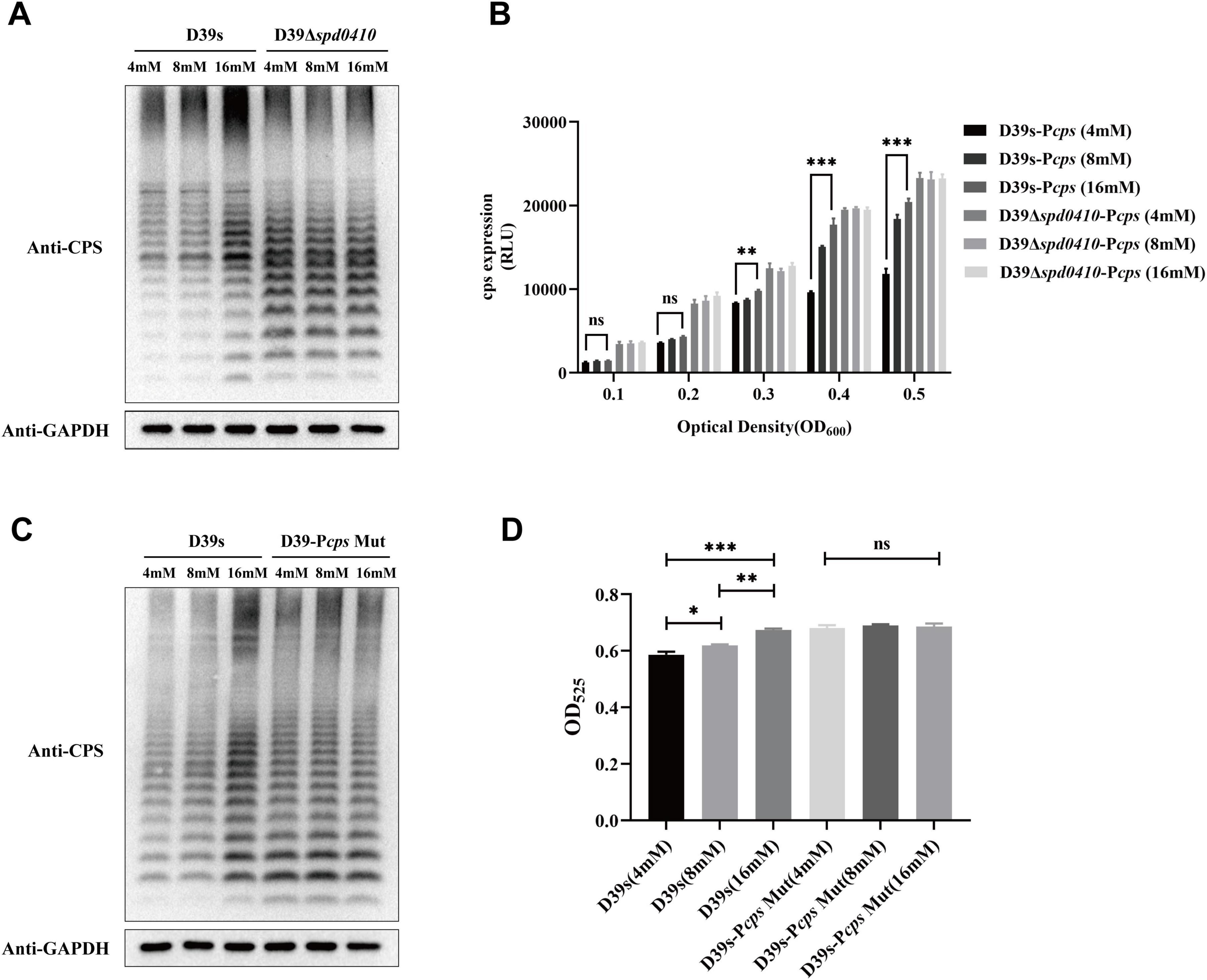
Figure 6. Glucose level affects CPS expression. (A) The expression of CPS in D39s and D39Δspd0410 strains at different glucose concentrations was detected by western blotting. (B) Luciferase activity analysis evaluated the expression of cps promoters in D39s-Pcps and D39Δspd0410-Pcps strains at different glucose concentrations. The results of representative experiments are presented as the mean of three replicates ± SD. ***P < 0.001; **P < 0.01; ns, not significant, as analyzed by as analyzed by unpaired two-way ANOVA. (C) The expression of CPS in D39s and each mutant strain in different glucose concentrations was detected by western blotting. (D) Determination of whole-cell CPS levels in D39s and D39Δspd0410 strains at different glucose concentrations. The results of representative experiments are presented as the mean of three replicates ± SD.***P < 0.001; **P < 0.01; *P < 0.05; ns, not significant, as analyzed by unpaired two-tailed Student’s t-test.
3.6 Deletion of spd0410 influences adhesion and pathogenicity of S. pneumoniaeDue to the pivotal role of the capsule in the pathogenesis of S. pneumoniae, we subsequently investigated the impact of the SPD_0410 on bacterial virulence. The capsule effectively aids the bacteria in evading phagocytosis by the host immune system, so we examined our test strains in phagocytic evasion assays using primary mouse peritoneal macrophages. We found that D39Δspd0410 exhibited increased resistance to phagocytosis compared to D39s (Figure 7A). Additionally, adhesion to A549 cells was decreased in the mutant D39Δspd0410 (Figure 7B). In contrast, the D39Δspd0410 strain demonstrated enhanced invasion capabilities and penetrated the A549 cells monolayers more effectively than D39s (Figure 7C).
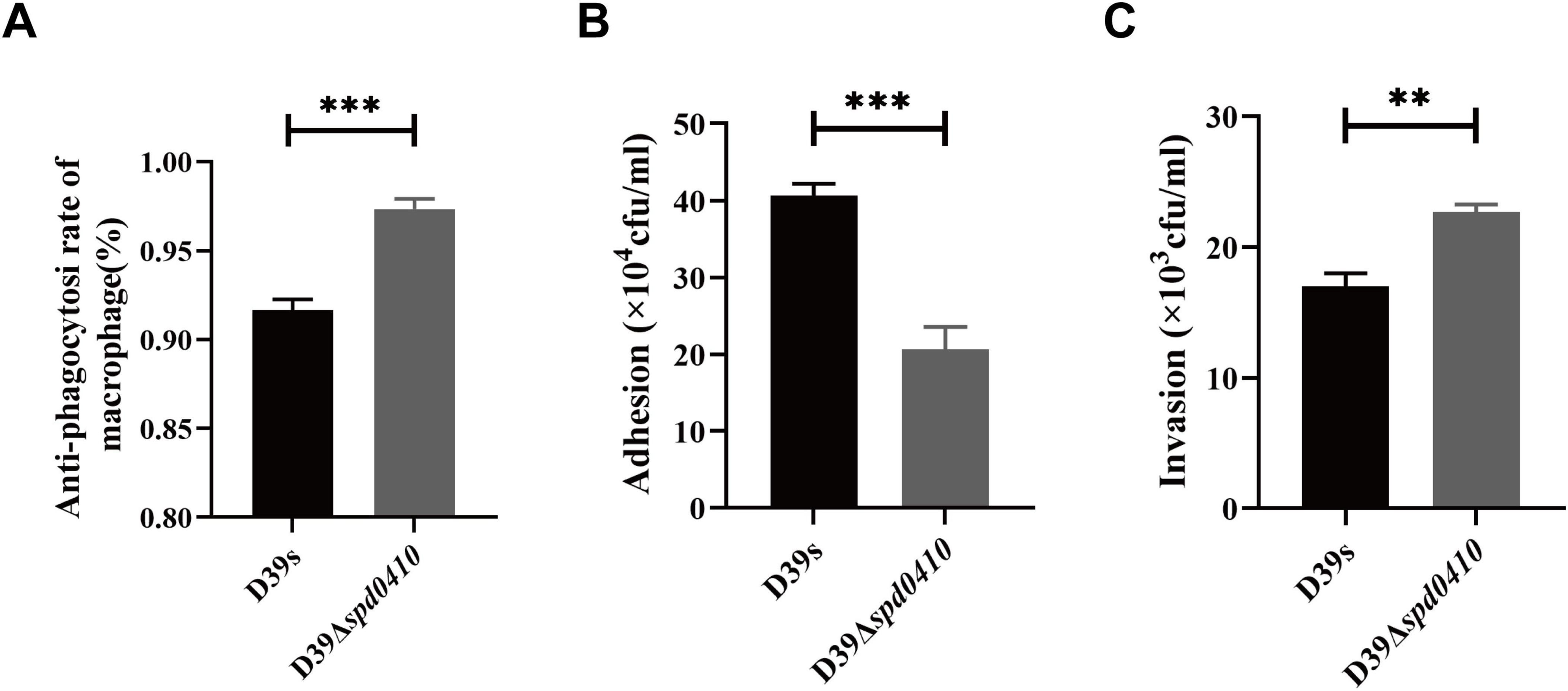
Figure 7. Macrophage phagocytosis and infection of epithelial cells. (A) Comparison of the ability between D39s and D39Δspd0410 against phagocytosis from mouse peritoneal primary macrophages. The cells were infected at an MOI of 100 with the indicated strains. (B,C) Infection of epithelial A549 cells. The cells were infected at an MOI of 100 with the indicated strains. (B) The number of bacterial adhesion to A549 cells. (C) The number of bacterial invasions to A549 cells. ***P < 0.001; **P < 0.01, analyzed by unpaired two-tailed Student’s t-test.
3.7 SPD_0410 is involved in systemic virulence and nasopharyngeal colonization of S. pneumoniaeTo thoroughly investigate the impact of SPD_0410 on the infectivity of S. pneumoniae, we performed an in vivo assessment of infection in a mouse model using intranasal administration of the bacteria to establish a pneumonia model. We found that mice infected with D39Δspd0410 had a significantly reduced survival compared to the parental strain D39s (Figure 8A). We further examined the invasion capabilities of D39s and D39Δspd0410 strains in the mice, observed pathological lung damage, and measured bacterial loads after 48 h. H&E staining revealed greater inflammatory cell infiltration and more pronounced pathological damage in the D39Δspd0410 infection group (Figure 8B). We also observed increased bacterial colonization in the lungs of mice infected with D39Δspd0410 (Figure 8C). CFU in the nasopharyngeal lavage solutions of D39Δspd0410-infected mice was lower than that of D39s-infected (Figure 8D), and this was consistent with the decreased adhesion levels for the mutant to lung epithelial cells (Figure 7B). In addition, mice infected with D39Δspd0410 showed greater susceptibility to invasion of the spleen and blood (Figures 8E, F).
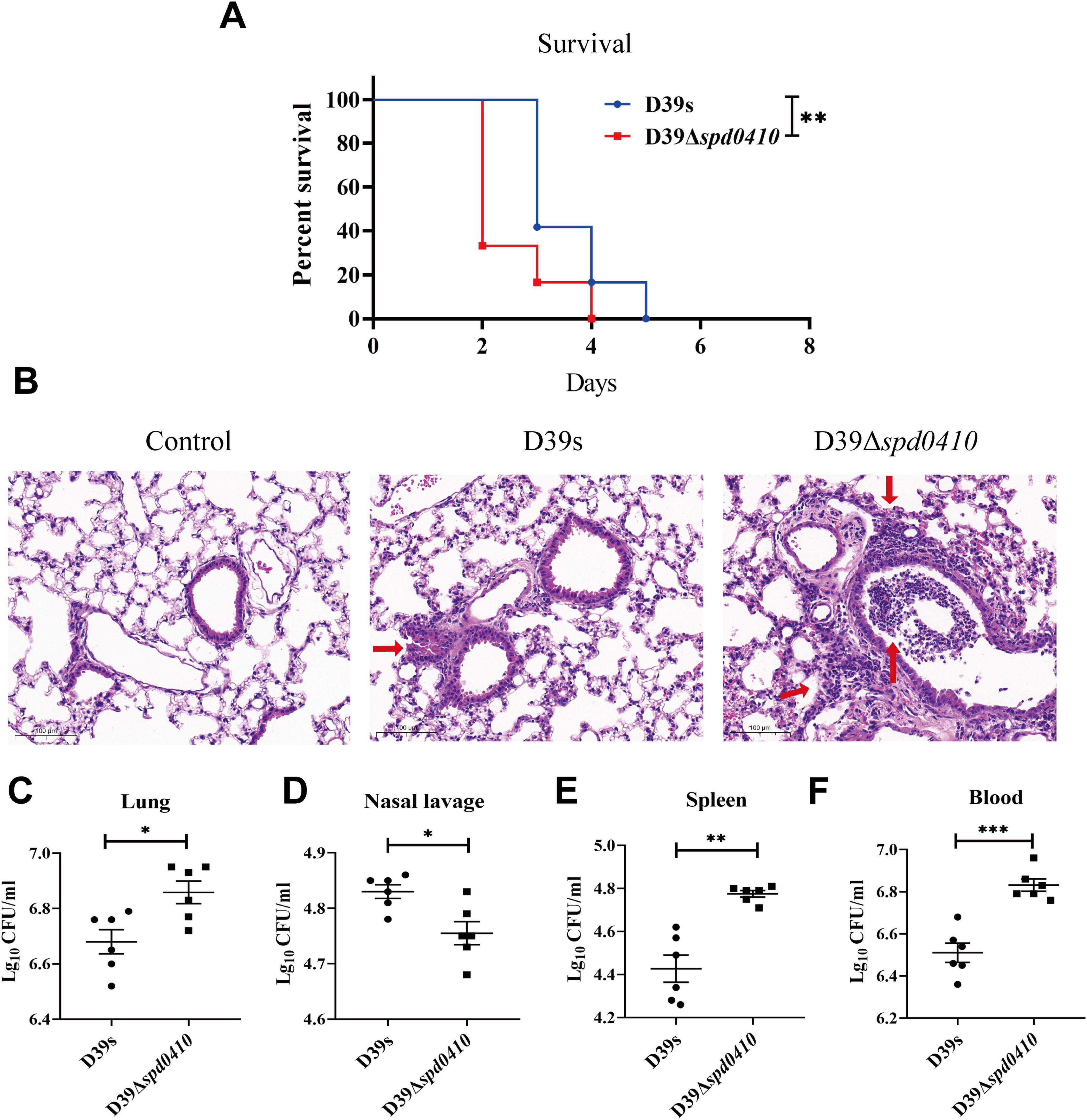
Figure 8. Survival of infected mice and bacterial colonization. (A) The survival curves of D39s and 39Δspd0410 strains infected mice via intranasal inoculation of 1 × 108 CFU S. pneumoniae into C57BL/6 mice (n = 12 animals per group). Survival was analyzed using log-rank comparisons. **P < 0.05. (B) Photomicrograph of H&E-stained lung tissues of mice 48 h after bacterial infection in mice at the same dose as the colonization experiments. Red arrows indicate the inflammatory cells. (C–F) Intranasal infection with 2 × 107 CFU of S. pneumoniae to observe bacterial colonization (n = 6 animals per group). Bacterial loads were evaluated by cultures of (C) lung homogenates, (D) nasal lavage, (E) spleen homogenates, and (F) blood. The results of representative experiments are presented as the mean of three replicates ± SD. ***P < 0.001; **P < 0.01; *P < 0.05, as analyzed by the non-parametric Mann–Whitney test.
Overall, these findings are consistent with the in vitro virulence experiments and indicate that SPD_0410 is a negative regulator of bacterial virulence that works by altering CPS levels on the bacterial cell surface.
4 DiscussionThe S. pneumoniae capsule is crucial for bacterial virulence and has historically provided the first example of CPS linkage to virulence as well as to the transfer of genetic information via DNA (Griffith, 1928). CPS synthesis occurs through two primary pathways: single sugar polymerization (synthase-dependent) and discrete repeat unit assembly (Wzx/Wzy-dependent) (Yother, 2011). The synthase-dependent pathway progressively adds sugar through a single enzyme and occurs in serotypes 3 and 37 (Engholm et al., 2017). Conversely, other serotype strains follow the Wzx/Wzy-dependent pathway, synthesizing repeat units by adding sugars to the inner layer of the cell membrane and transferring sugar chains to the outer layer by the Wzy polymerase (Whitfield et al., 2020). In the Wzy synthesis pathway, the cps gene forms an operon between two genes not involved in capsule biosynthesis; dexB, and aliA. The first four genes in the CPS operon (cpsABCD), are widely conserved but their exact functions have not been fully elucidated (Kolkman et al., 1998; Yother, 2011).
Previous studies have reported that the cpsA gene product CpsA acts as a transcriptional regulator of cps loci by specifically binding to the cps promoter sequences and this has been shown for both Streptococcus iniae and Streptococcus agalactiae (Hanson et al., 2010; Hanson et al., 2012). However, this conclusion has not been confirmed in the D39 strain. Additional studies have shown that CpsA is linked to CPS attachment to cell wall peptidoglycans (Stefanović et al., 2021). The expression of cps loci is regulated via specific transcription factor expression and responses to environmental signals (Xiao et al., 2021). Transcriptional regulation is essential for S. pneumoniae to adapt to changes in the host environment (Aprianto et al., 2018) and involves two-component signal transduction systems (Standish et al., 2005; Yu et al., 2023). A previous study had determined that mRNA levels from the cps locus in strain D39 were upregulated in the bloodstream during pneumococcal infection (Ogunniyi et al., 2002). Together, these data suggest that regulation of cps transcription can be influenced by environmental conditions.
In previous studies, we identified that the protein SPD_0410 can bind to the cps promoter in DNA pull-down experiments and this suggested its potential involvement in CPS synthesis (Wu et al., 2016). In the current study, we focused on whether the SPD_0410 protein is a regulator of CPS. We observed that the absence of spd0410 led to increased CPS levels on the bacterial surface and inside the cells implicating this protein as a negative regulator. EMSA and DNase I footprinting analyses confirmed that SPD_0410 specifically binds to two distinct sites within the cps promoter region and therefore functions as a novel factor that can regulate the cps gene locus. However, surprisingly, our transcriptome sequencing did not reveal significant changes for the cps gene locus and this may be attributed to samples taken from the logarithmic growth phase and agrees with the qRT-PCR findings. Since we found significant upregulation of mRNA levels from the cps locus in the early stages of growth in D39Δspd0410, we suspect that SPD_0410 may be responding to new environmental or nutritional changes in the early stages of growth. Thus, these factors would trigger specific regulatory mechanisms of the cps gene clusters. However, this regulation may be adjusted or alleviated as bacteria progress into the logarithmic growth phase. Nevertheless, we consider this data to be significant as it highlights the complexity of bacterial physiology.
Previous studies have indicated that a group of cps transcriptional activators typically possess conserved domains that enable them to function as transcription factors, particularly as global transcriptional regulators (Moscoso and García, 2009; Solano-Collado et al., 2016). However, our comparison of the amino acid sequence of SPD_0410 revealed that its orthologs were hypothetical proteins with no detailed reports on their biological functions. Surprisingly, we found that SPD_0410 in S. pneumoniae exhibits a certain similarity to its homologous protein containing a helix-turn-helix structure. This suggests that SPD_0410 may also bind to specific DNA sites in a dimeric form. This hypothesis is supported by our gel shift and footprinting experiments that demonstrated SPD_0410 binding to the cps promoter at two specific sites. These findings provide insights for the further investigation of the complete biological function of SPD_0410.
The growth of S. pneumoniae is governed by carbon catabolite repression via the transcriptional regulator CcpA. As such, the carbon source influences capsule synthesis and thickness across different serotypes (Iyer et al., 2005; Gö
留言 (0)