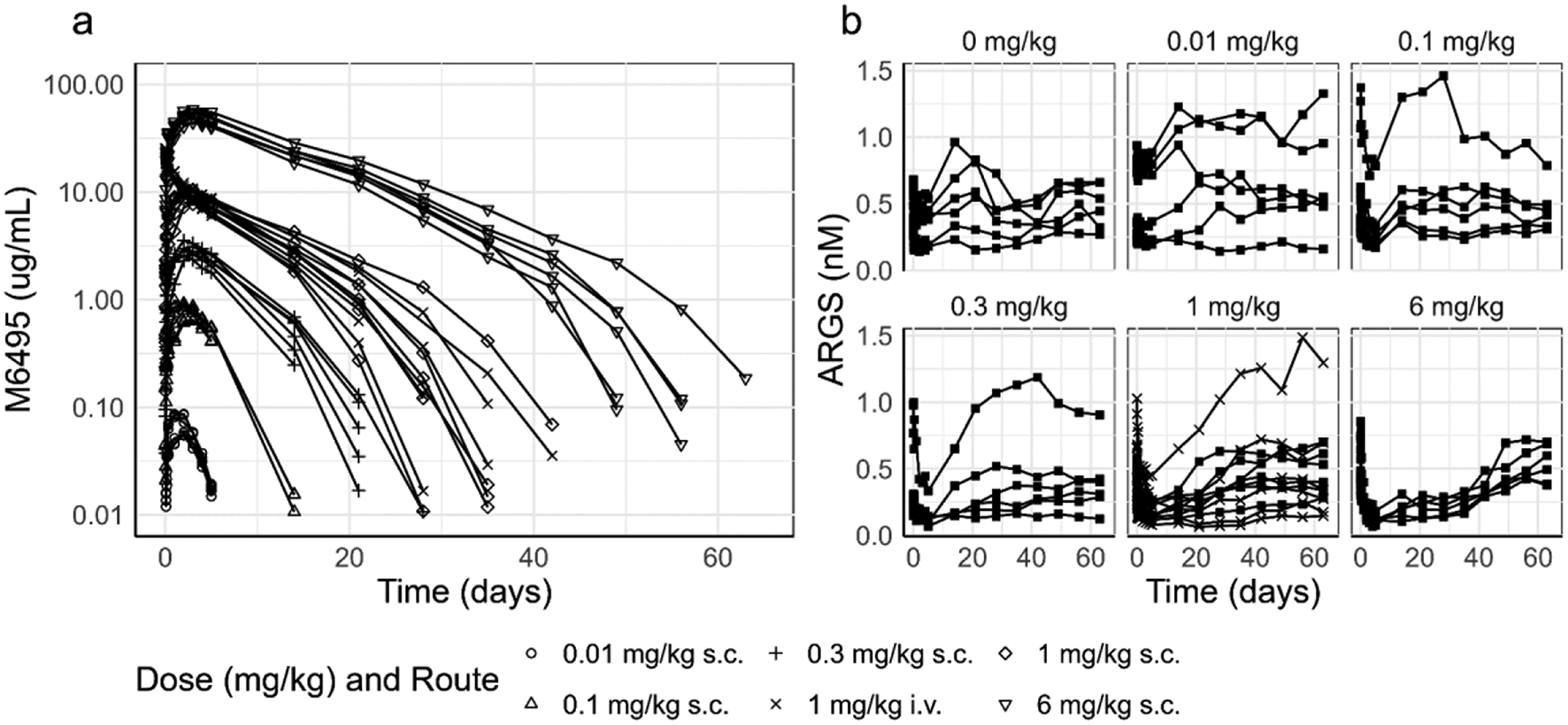
M6495 pharmacokinetics and ARGS modulation in cynomolgus monkeys
All in vivo studies were performed in accordance with the local guidelines of the Institutional Animal Care and Use Committee. Two studies were performed in cynomolgus monkeys, a single dose PK/PD study to study M6495 pharmacokinetics and pharmacodynamics and a single and multiple-dose safety study, where the biomarker ARGS was also measured. The main route of administration was s.c., and an i.v. group (1 mg/kg) was included to assess the absolute bioavailability of M6495. PK and PD samples were taken up to 672 h (28 days) in the multiple dose study and up to 1512 h (63 days) in the single dose PK/PD study. A summary of study design, doses and sample size is presented in Supplementary Tables 1 and 2. All samples were analyzed for M6495 and ARGS using immunoassay sandwich methods. The lower limit of quantification (LLOQ) for the detection of M6495 in serum was 3.2ng/mL as determined using qualified assay and validated assays.
As described in the introduction, the neo-epitope ARGS, which is a product of the cleavage of aggrecan by M6495, was used as a pharmacodynamic marker. A ligand binding assay was used for the detection of the concentrations of ARGS in serum. This assay employs a biotinylated mAb (monoclonal antibody) targeting the G2 domain of aggrecan as a capture reagent, and a horse-radish peroxidase conjugated mAb targeting the ARGS neo-epitope as detection reagent (the assays were developed and validated at Nordic Bioscience, Denmark). The assay used for assessing ARGS concentrations in the single and multiple-dose safety was characterized with a lower and an upper limit of quantification of 0.08 nM and 1.8 nM, respectively. This assay was further analytically validated in support of the PK/PD single dose study, resulting in a LLOQ (lower limit of quantification) of 0.064 and a ULOQ (upper limit of quantification) of 0.5 nM. Samples above the quantitative range were diluted into the quantitative range, supported by the dilution recovery (parallelism) evaluation of the assay as assessed during the analytical validation, and the parallelism was considered fit-for-purpose.
Pharmacokinetic and pharmacodynamic data from the single dose PK/PD study and the safety study in cynomolgus monkeys were pooled for the population PK/PD model building using nonlinear mixed effect methods. Data below the LLOQ were treated as missing in the modelling dataset, or set to 0 for graphical presentation.
Modelling of M6495 PK/PD in cynomolgus monkeys
Nonlinear mixed-effects modelling was performed using NONMEM 7.3 (ICON Development Solutions) and the software tool PsN version 4.6.0. The data and various model outcomes of the NONMEM model outputs were explored graphically using R version 3.3.1.
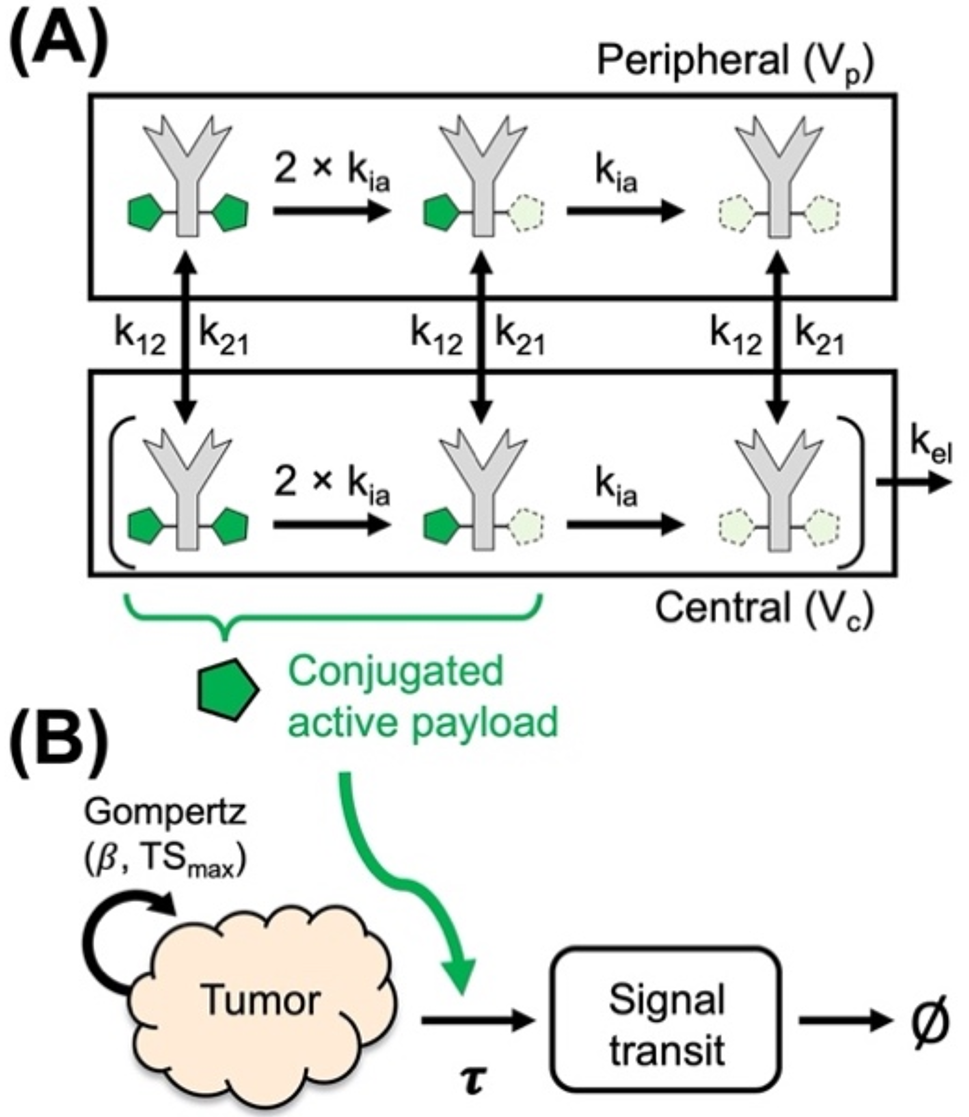
A PK model was developed based on the subcutaneous data, and the intravenous administration data were added to the dataset allowing the estimation of the bioavailability. This PK model was subsequently used as a starting point for simultaneously fitting the PK (M6495) and PD (ARGS) data.
For describing M6495 pharmacokinetics, a 2-compartment model with parallel linear and non-linear clearance was used, as described by the following differential equations (1 to 4):
$$\:Kel=\frac;k12=\frac;k21=\frac\:;\:\text1\:=\:\text1/\text1\:\:$$
(1)
$$\:\frac=-Ka*A\:\:$$
(2)
$$\eqalign}1} \over } = Ka*F*A - k12*}1 + k21*}2 \cr & - kel}1 - Vmax*}1} \over }1}}} \right)} \over }1} \over }1}}} \right)}} \cr}$$
(3)
$$\:\frac=k12*A1-k21*A2$$
(4)
The differential equations for the modelling in NONMEM were parameterized with A, A1 and A2, representing the amounts in the depot compartment and in the central and peripheral compartments, respectively, and with micro-constants kel, k12 and k21, described in the Eq. 1. The parameters ka, F, Q, CL, Vmax and Km represent the absorption rate constant, bioavailability, inter-compartmental flow, clearance, maximal rate of non-linear clearance and the concentration at which the non-linear clearance is 50% of the maximum value, respectively. The Michaelis Menten parameter Km was described in concentration units, and the equation expressed in amount divided by the volume of distribution (A1/V1, Eq. 3). The concentration of M6495 in the central compartment (C1 = A1/V1) was fitted to the PK data.
The pharmacokinetics and pharmacodynamics of M6495 were modelled simultaneously. M6495 pharmacodynamics were described with an indirect response model as illustrated by Eq. (5), where ARGS is the concentration of ARGS measured in serum in nM. The parameters kin, kout, Imax and IC50 represent, respectively, the synthesis rate of ARGS, the elimination rate constant of ARGS, the maximal inhibition of ARGS synthesis and the concentration at which 50% of the maximal inhibition is achieved.
$$\:\frac=kin\:\left(\:1-\frac\right)-kout*ARGS$$
(5)
The synthesis rate of ARGS was defined as described by Eq. 6, assuming steady state at time = 0, where ARGSbaseline is the baseline concentration of ARGS, which is a model parameter.
$$\:kin=ARG_*kout\:\:$$
(6)
Also at time = 0, as initial condition for the model,
The first-order conditional estimation with η-ε interaction in NONMEM 7.3 was employed for all model runs. Inter-individual variability on the parameters was expressed as an exponential error model based on the assumption of a log-normal distribution. Imax was parameterized using a box-cox transformation (Eq. 8 to 9) on the interindividual variability parameter (η) for Imax (ηImax) in order to restrict the value to between 0 and 1 [10]. Imax was therefore defined by its typical population value, ϴImax, and the transformed η (Eq. 10).
$$\Phi = }^\nolimits} }}}}$$
(8)
$$}}} = }}} - 1} \over }}}$$
(9)
$$\nolimits} = \nolimits} }}*}}}}}$$
(10)
The model validation was based on graphical analyses and a prediction corrected visual predictive check shown in Supplementary Fig. 2 to 5 (pcVPC). Assessment of model adequacy was driven by the data and guided by goodness of fit criteria [11]. Parameter estimates were reported with a measure of estimation uncertainty, the standard error of the estimates.
Predicting M6495 human pharmacokinetics and pharmacodynamics
The parameters obtained from the model in cynomolgus monkey were used to predict M6495 pharmacokinetics and pharmacodynamics in humans after subcutaneous administration of M6495 at different dose levels, and to anticipate the possible doses in human based on the reduction of ARGS levels. The Pharmacokinetic parameters V1, V2, Q, CL, Vmax and Ka were scaled allometrically from the cynomolgus monkey model according to the following relationship, using the exponents 1, 1, 0.75, 0.75, 0.75 and − 0.25, respectively:
$$\:Paramete_=_*_}_}\right)}^\:\:$$
(11)
The parameters F, Km, Imax, IC50 and kout were assumed to be the same as in the cynomolgus monkey model. The baseline ARGS concentration parameter (ARGSbaseline) in the cynomolgus monkey model was replaced by data generated in-house for the ARGS concentration in human serum and respective variability (0.14 nM, 46% CV), which were available from serum samples from 48 healthy humans. These scaling assumptions are summarized in Table 2 and in more detail in Supplementary Table 3. The exponential interindividual variability in the linear PK parameters CL and Ka was increased to a variance of 0.1 to account for the higher pharmacokinetic variability anticipated in a human clinical trial (this would correspond to ~ 30% interindividual variability). A similar approach of inflating variability in clearance to predict human pharmacokinetics was suggested and used successfully by Deng et al. [12]. The interindividual variability in the parameter accounting for non-linear clearance Vmax was kept the same as in the cynomolgus monkey. The interindividual variability in the pharmacodynamic parameter Imax was assumed to be the same as in the cynomolgus monkey, while the ARGS baseline parameter was replaced by own data, as described above.
The pharmacokinetic and pharmacodynamic profiles for 500 virtual subjects were simulated based on the previously described model and scaled parameters, (Fig. 4).
The maximal individual decrease in the ARGS biomarker simulated through the human scaled model was calculated as percentage from baseline and summarized with descriptive statistics over the simulated dose range (Fig. 5). While ARGS levels expressed as concentration values (nM) was appropriate for both the monkey PK/PD model and the human predictions, the values were subsequently converted to percentage decrease from baseline in order to simulate the dose range for the human Phase I trial, and to allow comparison with the metric used to evaluate ARGS in the human trial [4].
The simulations of the scaled PK/PD model for predicting human PKPD shown in this publication were performed in R version 4.1.0 using the RxODE package (1.0.9), and the code is included as a supplementary information for reproducibility in an open-source format, and for further use for research in this field.






留言 (0)