
記住我




The SPIRIT reporting guidelines and SPIRIT figure were used to create this manuscript [31]. Our methods section closely resembles but is not identical to that of the published protocol on AB-free kava’s effect on decreasing tobacco-associated lung cancer risk [32].
Study settingThe study will take place within clinics of University of Florida (UF) Health Family Medicine. Clinic staff members from the UF Health Family Medicine Main Street Clinic and Springhill Clinic, under the supervision of medical doctors, will identify possible participants and introduce the overall goal of the study, typically during the regular clinic visit of the prospective participants.
Study setting justificationUF Health Family Medicine Main Street Clinic and Springhill sites are chosen for the following reasons: (a) over 1700 adult smokers visited the Main Street Clinic last year for annual checkup, > 50% of whom smoke at least 10 cigarettes/day. Although our exclusion criteria are stringent (detailed below), we expect at least 15–20% recruitment rate given the high success to recruit participants in family clinics for tobacco trials; (b) successful track record of recruiting smokers to support clinical research, including collaborations between our co-investigators who have previously recruited 120 adult smokers from the same clinic in 4.5 months (NCT03836573); (c) electronic health record information of patients in the clinics will help identify eligible participants – those without previous liver diseases; and (d) most importantly, the clinical expertise, resources, and facilities will minimize potential adverse effects in this trial. The Springhill site has a similar patient population and clinical research structure. We propose two sites to maximize enrollment.
Eligibility criteriaIn this study, we have utilized the same eligibility criteria as our previously published paper “Reducing tobacco-associated lung cancer risk: a study protocol for a randomized clinical trial of AB-free kava” with one notable exception: participants are now required to express a desire to quit. This modification reflects an evolving emphasis on participant motivation and readiness to engage in cessation efforts, which was not a prerequisite in our earlier investigation. By incorporating this criterion, we aim to enhance the relevance and applicability of our findings to current clinical practices. This adjustment ensures that our study maintains methodological rigor while accommodating advancements in the field, ultimately contributing to a more nuanced understanding of smoking cessation interventions.
By adhering to established criteria, we aimed to maintain methodological continuity and facilitate direct comparisons with previous findings. This approach not only strengthens the coherence of our study design but also underscores the reliability and validity of our results within the context of our ongoing research program.
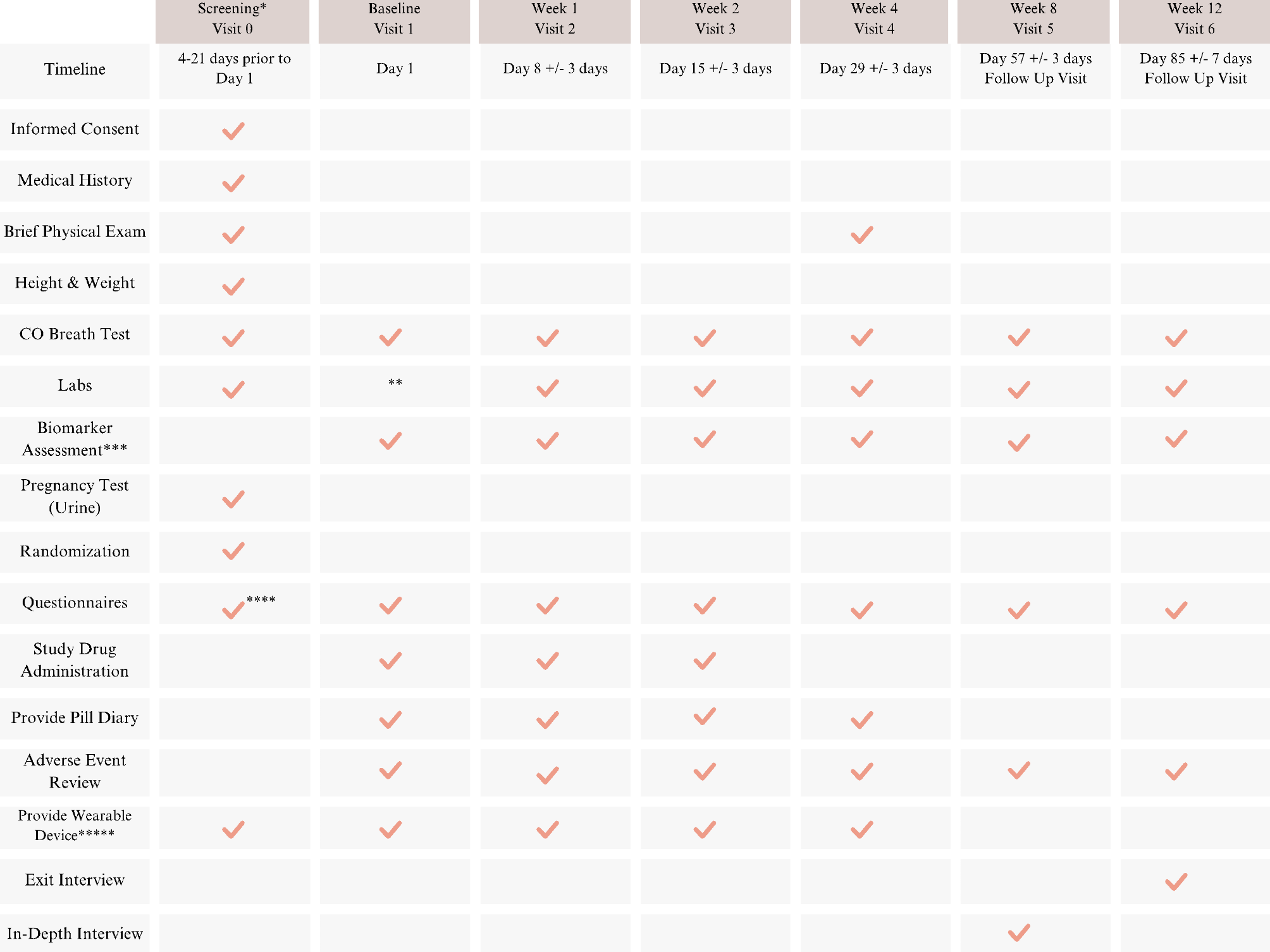
Intervention descriptionThe trial will require resources from both the UF Family Medicine Clinics and UF Consent2Share to assist in the recruitment of active smokers intending to quit, who are otherwise healthy. These participants will be assigned randomly to either the placebo group or the experimental group receiving AB-free kava (225 mg dose), undergoing a four-week exposure period with a total of six visits (Week 0, 1, 2, 4, 8, and 12). The primary objectives of these visits are to assess adherence and pinpoint any potential challenges related to the use of AB-free kava. Moreover, this study seeks to investigate how AB-free kava affects tobacco use and dependence, stress levels, sleep patterns, and associated indicators among study enrollees.
A total of three capsules of placebo or AB-free kava is administered by mouth daily. Participants will be instructed to take one capsule in the morning, at noon, and in the evening, at approximately the same time each day. Each AB-free kava capsule contains 75 mg of kavalactones, resulting in daily regimen of 225 mg. The dosage will remain unmodified, and in case of any adverse effect (detailed in a later section), usage will be stopped. Permission to conduct this study will be obtained from the UF IRB prior to start. The content of each visit is outlined in the participant timeline depicted in Fig. 1.
Potential participants will be identified and referred to the study coordinators by the clinic staff members. Upon referral, the coordinator will conduct the pre-screening interview. During the interview, the study and its potential benefits and risks will be explained. The study coordinator will schedule interested candidates that meet eligibility criteria for a pre-screening visit. At that visit, study coordinators will facilitate the process of informed consent for potential participants. This involves ensuring individuals receive comprehensive information about the study, and their rights as participants. The study coordinator will also emphasize the importance of limiting alcohol consumption and avoiding any medications containing acetaminophen. Participants will then verbalize understanding that their participation is voluntary, and they have the option to withdraw without facing implications regarding their current or future healthcare. They will also be made aware that they may be withdrawn by the study team if they exhibit non-adherence to the protocol, fail to limit alcohol intake, or experience adverse events, among other reasons explained later.
After allowing them to review the informed consent and ask any questions, they will sign the consent form electronically in the highly secure and HIPAA-compliant web-based application, Research Electronic Data Capture (REDCap). Study coordinators will inform the candidates that they will receive a $50 gift card/visit at Visit 1–6 that will total up to $300 if they attend all visits. This $300 amount does not include the optional $25 that selected subjects may receive for consenting to complete the post-treatment interview at Visit 4 (or whenever they stop treatment). If subjects complete all visits and do the optional post-treatment interview, then they can receive a total of $325.
Participants will then take a Carbon Monoxide (CO) breath test to assess smoking status. Blood will be collected to determine whether ALT, AST, ALP, and total bilirubin levels fall within normal ranges. A pregnancy spot urine test will also be done for females of childbearing potential. Once eligibility criteria are confirmed, qualified participants will be enrolled into this study and assigned randomly to either of the study groups. Visit 1 will also be set up within 4 to 21 days later. The research coordinator will provide the participant with a GT3X + accelerometer wearable device and will instruct them to wear it for seven days after the first three visits at Visit 0, Visit 1, and Visit 2. The research coordinator will also instruct participant to wear the device 7 days prior to Visit 4. The wearable device will use a micro-electro-mechanical system (MEMS) based accelerometer and an ambient light sensor to measure physical activity and sleep. The device will be programmed to begin recording at midnight after their visit. The study coordinator will provide instruction on the use of the wearable device, including demonstration on how to correctly wear the device.
Each of the six visits, following the screening visit, will last around 30–60 min. These visits will involve blood tests for ALT, AST, ALP, total bilirubin levels, a breath CO test, and the administration of questionnaires. Figure 2 details the contents of the Comprehensive Metabolic Panel lab test along with their respective units. A10 mL blood sample will also be collected onsite, along with a 24-hour urine collection, for biomarker analysis. The participants will also be given the medication in a bottle that has written instructions and a pill diary to keep track of their medication doses and note the corresponding times. This tool will be useful for comparing the number of pills returned in the bottle to the information recorded in the pill diary. Additionally, they will be provided with materials for the upcoming urine collection. Participants will have a continuous access to the study coordinator, fostering a strong rapport to encourage their return. Additionally, they will be closely monitored throughout their participation, and study visits will be confirmed via reminder phone calls. Upon completion of each visit, participants will receive a debit card loaded with funds, which will be reloaded at every subsequent visit. The study coordinators will consistently employ respectful language and demonstrate empathy when communicating with the participants.
During Visit 1–4, the data stored on the GT3X + wearable device will be downloaded by the study coordinator. At Visit 4, the GT3X + wearable device will be collected from the participants.
At the conclusion of the study (Visit 6) or upon a participant’s withdrawal from the study, they will undergo an exit interview consisting of open-ended questions to gather their feedback on the trial.
At the end of the treatment, Visit 4, or at the time a participant withdraws from the study, some participants will be offered an opportunity to complete a post-treatment interview. If they choose to participate, they will receive an additional $25 to the $50 they are already being given for their attendance at that visit. Participants who are invited to complete the post-treatment interview will be purposively sought to ensure comparable subgroups of racial and ethnic minorities and white participants. By recruiting these subgroups, we can explore cultural similarities and differences in their experiences. We expect to recruit a total of 25 participants to complete the post-treatment interview to ensure thematic saturation. However, data collection and analysis will be concurrent to ensure saturation is reached. This interview will help to identify facilitators and barriers to trial enrollment and retention.
Due to the established rapport and trust, the study coordinator will conduct thorough, semi-structured interviews either via phone or Zoom [33]. Dr. Fisher (co-I), a qualitative methodology expert in health behavior intervention development, will provide oversight on data collection and analysis. She will also develop the semi-structured script and train the coordinator on in-depth interviewing. Interview questions will explore participants’ experiences with kava to identify critical factors to promoting future trial enrollment, adherence, and sustained healthy behavior change [34, 35]. Questions will explore facilitators and barriers to enrollment and retention as well as sociocultural factors informing participants’ health behavior and ongoing willingness to engage in kava use. These include exploring the influential role of cultural norms or beliefs, experiences within their family system, and prior use or beliefs about kava. The research coordinator will audio record the interviews and will send the files to be transcribed professionally. The transcriptions of interviews are the qualitative data that will be thematically analyzed by a qualitative research scientist. Without audio files or transcriptions, there will be no reliable data. This approach conforms with standard and best practices in qualitative methodology.
OutcomesThe primary endpoint is to evaluate participants’ compliance to AB-free kava. Such information is critical for data interpretation and future clinical trial improvement. We will evaluate trial compliance using the same three methods employed during the pilot trial: [30] (i) participants will report any missed doses via their pill diaries; (ii) the UF IDS pharmacy will count returned pills; and (iii) and the study team will analyze participants’ urine for the presence of dihydromethysticin (DHM) during kava ingestion, as DHM is specific to kava [36]. In combination with the exit interview, the results will provide knowledge of the practicality of using AB-free kava and potential issues with current treatment regimen among smokers.
Another study aim is to investigate if AB-free kava can help patients quit smoking, reduce stress, and enhance sleep quality. Tobacco dependence will be determined using questionnaires, inquiring about participants’ smoking urges. Tobacco use will be assessed using measurement of urinary TNE. Stress reduction will be studied using perceived stress questions and stress biomarkers such as plasma cortisol, PRKACA, and urinary TCE. Sleep improvement will be determined using sleep quality questionnaires, wearable accelerometer measurements, and sleep biomarkers (urinary 6-hydroxymelatonin and urinary NAS). Assessments will be made on the variations in participants’ results and responses in comparison to their baseline data.
Participant timelineFig. 1
Participant timeline that includes an AB-free Kava intervention, followed by follow-up sessions. Legend: *: Screening procedures conducted according to established medical standards during the designated screening period can be administered before formal consent is obtained. **: If the baseline visit is within 7 days of the screening, there is no need to redraw blood. Labs are listed in the table below. ***: Blood and urine markers. ****: Only ASQ questionnaire is administered at screening. *****: GTR3X + will be provided to participants. The participants will wear the device for 7 days after visits 0, 1, and 2, and 7 days prior to visit. They will also be instructed to refrain from wearing the device following visit 3
Fig. 2 Sample size
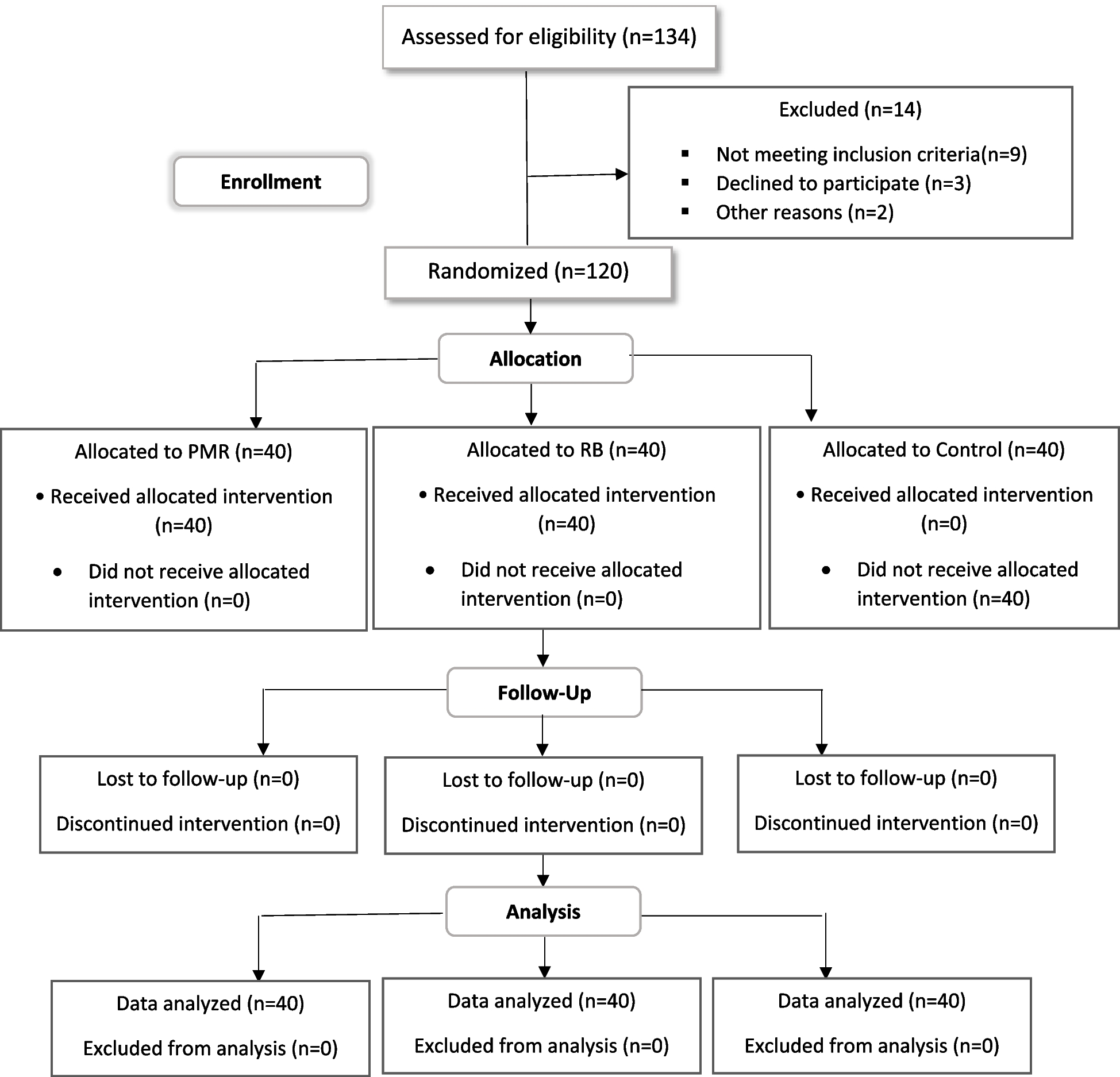
Sample sizeOur targeted accrual goal is 76 active smokers. Based on our previous enrolling experience at the UF Health Family Medicine Clinic and our rather strict eligibility criteria, the enrollment is expected to be 1.5–2.5 years with the study duration of each participant to be approximately 12 weeks.
The sample size was determined based on our primary biological signature endpoint data (TNE and PRKACA) from our pilot results. Using the pilot result of TNE as an example, the ratio of geometric mean of TNE after one week kava treatment is 0.68 with a standard error of 0.06. By taking into consideration that the placebo group demonstrates 15% of the treatment impact, a figure widely agreed upon in existing literature [37, 38], and apply the identical standard error as observed in the kava treatment group, the standardized effect size (Cohen’s d) equals d = 1.01. Similarly, the Cohen’s d for PRKACA is 0.89. Since in our proposed study, we will repeatedly measure these biomarkers in each participant, we further assume the same Kava effect from week one through week four. By (i) varying within-participant correlation rho = 0, 0.2, 0.4, 0.6, and 0.8; (ii) at Bonferroni-corrected alpha level 2.5% (5%/2), and (iii) using a linear mixed model, 60 participants (30 per group) are needed to have at least 90% power for both TNE and PRKACA. Additionally, supposing an 80% retention rate, consistent with rates in comparable studies [39], tour plan entails recruiting 76 participants, with 38 assigned to each group.
The research team will exclude pregnant women from this study due to the proposed method involving AB-free kava to help reduce tobacco use by managing stress. This decision is based on the anticipation that pregnancy may induce substantial alterations in mental state and hormonal physiology. These discrepancies are likely to impede the primary objective of this early-phase clinical investigation by weakening the justification for determining the sample size.
RecruitmentWe intend to reach our target sample size through several strategies. Firstly, we will engage the clinic staff, ensuring they are well-informed about the study and addressing any questions they may have. Secondly, we will utilize UF Consent2Share to disseminate our study and connect with a larger number of patients who visit UF Health clinics. Additionally, consistent reminders and effective communication with clinic staff will be maintained throughout the study. Flyers promoting the study will be distributed at the clinic site to motivate patients to participate. Our study coordinators will focus on establishing strong rapport with patients to enhance retention rates. Lastly, participants’ involvement will be compensated through the distribution of debit cards.
留言 (0)