
記住我
Dissimilation of glucose to carbon dioxide results in the generation of reducing equivalents, the primary type being in the form of NADH. For ongoing cell function, it is essential these are continuously (re)oxidized, a requirement intrinsically linked to the formation of ATP by the respiratory chain. In eukaryotes, glucose dissimilation and most of the NADH generation occurs in the cytosol during glycolysis and in the mitochondrial matrix by the combined action of the pyruvate-dehydrogenase complex and the tricarboxylic acid cycle. NADH cannot cross the inner mitochondrial membrane (von Jagow and Klingenberg, 1970), and as such needs to be (re)oxidized in the cellular compartment in which it was generated.
Fungal respiratory chains are typically branched and contain multiple entry points for electrons from NADH. While the large multi-subunit trans-membrane spanning type I NADH:quinone oxidoreductase (NDH-1 or Complex I) couples oxidation of mitochondrial NADH to the translocation of protons over the inner mitochondrial membrane, most fungi also possess monotopic alternative, type-II NADH dehydrogenases (NDH-2) that catalyze NADH:quinone oxidoreduction without proton translocation (Joseph-Horne et al., 2001). These ‘alternative NADH dehydrogenases’ are either peripheral membrane proteins, or monotopic membrane proteins (Boes et al., 2021) that are either associated or have a membrane anchoring helix attaching them to the inner mitochondrial membrane. They may differ in which side of the membrane they are located depending on cellular function. Their catalytic sites either face the mitochondrial matrix (‘internal’) where their catalytic activity overlaps with Complex I, or the intermembrane space (‘external’), allowing direct oxidation of cytosolic NADH (Antos-Krzeminska and Jarmuszkiewicz, 2019; Feng et al., 2012; Melo et al., 2004). Saccharomyces cerevisiae is an excellent example of an organism reliant on NDH-2. S. cerevisiae does not harbor a Complex I and relies solely on one internal and two external alternative NADH dehydrogenases as entry points for NADH-derived electrons into the respiratory chain (de Vries and Marres, 1987; Luttik et al., 1998). Besides NADH, some external fungal NDH-2, such as those from Kluyveromyces lactis (Tarrio et al., 2005; Tarrio et al., 2006) and Neurospora crassa (Carneiro et al., 2004; Carneiro et al., 2007; Melo et al., 2001), have also been reported to accept NADPH as substrate, either exclusively, or in addition to NADH. Based on the distribution of a key acidic residue (E272 in Ndi1 from S. cerevisiae), which has been proposed to prevent interaction with the phosphate group of NADPH (Iwata et al., 2012; Michalecka et al., 2004), it has been suggested that the majority of NDH-2 oxidize NADH rather than NADPH (Marreiros et al., 2016).
Ogataea parapolymorpha (formerly Hansenula polymorpha) is a methylotrophic, thermotolerant, Crabtree-negative yeast that is characterized by its rapid aerobic growth (Levine and Cooney, 1973; Juergens et al., 2018). It has a branched respiratory chain that contains one type I NADH dehydrogenase (Ndh1) and three putative type-II NAD(P)H dehydrogenases (referred to as Ndh2-1, Ndh2-2 and Ndh2-3 in this study) with unknown substrate specificity and unknown orientation on the inner mitochondrial membrane (Juergens et al., 2020). Interestingly, in the presence of excess glucose and oxygen, removal of Ndh1 from O. parapolymorpha does not result in a reduction of the maximum specific growth rate or biomass yield in batch cultures (Juergens et al., 2020). This implies that Ndh1 is disposable under these conditions. In aerobic glucose-limited cultures, elimination of Ndh1 resulted in mutant strain which exhibited a 16% lower biomass yield while it maintained a fully respiratory metabolism (Juergens et al., 2020). These phenotypes suggested that O. parapolymorpha harbors at least one internal alternative NADH dehydrogenase capable of compensating for loss of Ndh1, albeit at a lower efficiency of respiration-coupled ATP production due to a lack of protons pumped. Such an internal NADH dehydrogenase also would likely be responsible for (re)oxidation of mitochondrial NADH under fast-growing glucose-excess conditions, especially since NDH-2 appear to be suited to catalyze the high rates of NAD+ regeneration required to sustain a high glycolytic flux in the absence of fermentation (Godoy-Hernandez et al., 2019).
The aim of this study was to investigate the physiological roles of the three putative alternative NAD(P)H dehydrogenases of O. parapolymorpha with a special focus on determining whether this yeast indeed possesses an internal alternative NADH dehydrogenase capable of functionally substituting Ndh1. To this end, the aerobic growth characteristics of various O. parapolymorpha NAD(P)H-dehydrogenase mutant strains (Δndh2-1, Δndh2-2 and Δndh2-3) were investigated in glucose grown batch and chemostat cultures. To determine localization and substrate specificity of the alternative dehydrogenases, substrate-dependent oxygen-uptake rates of isolated mitochondria from wild type and mutant strains were measured, and activity of the individual membrane-bound dehydrogenases with NADH and NADPH was assessed.
Results Disruption of Ndh2-1 leads to a reduction of specific growth rate and a Crabtree-positive phenotype in Ogataea parapolymorphaTo investigate the contribution to respiration of the three putative alternative NAD(P)H dehydrogenases, strains IMD003, IMD004 and IMD005 were constructed, which harbored a disrupted version of the structural gene for Ndh2-1, Ndh2-2 or Ndh2-3, respectively (Δndh2-1, Δndh2-2 and Δndh2-3). Ndh2-1 is a peripheral membrane protein, while Ndh2-2 or Ndh2-3 are monotopic membrane proteins containing a single predicted transmembrane helix (see Supplementary Figure S1). The physiology of these mutant strains was then assessed in aerobic shake-flask batch cultures in the presence of excess glucose (2 g L−1). The high specific rate of oxygen uptake by Crabtree-negative yeasts can lead to oxygen limitation in shake-flask cultures resulting in respiro-fermentative metabolism (Kiers et al., 1998). However, under the cultivation conditions in this study, the wild-type O. parapolymorpha strain CBS11895 grows with a full respiratory phenotype. It has a comparable specific growth rate as previously described in fully aerobic bioreactors, indicating that oxygen limitation did not occur (Table 1; Juergens et al., 2018).
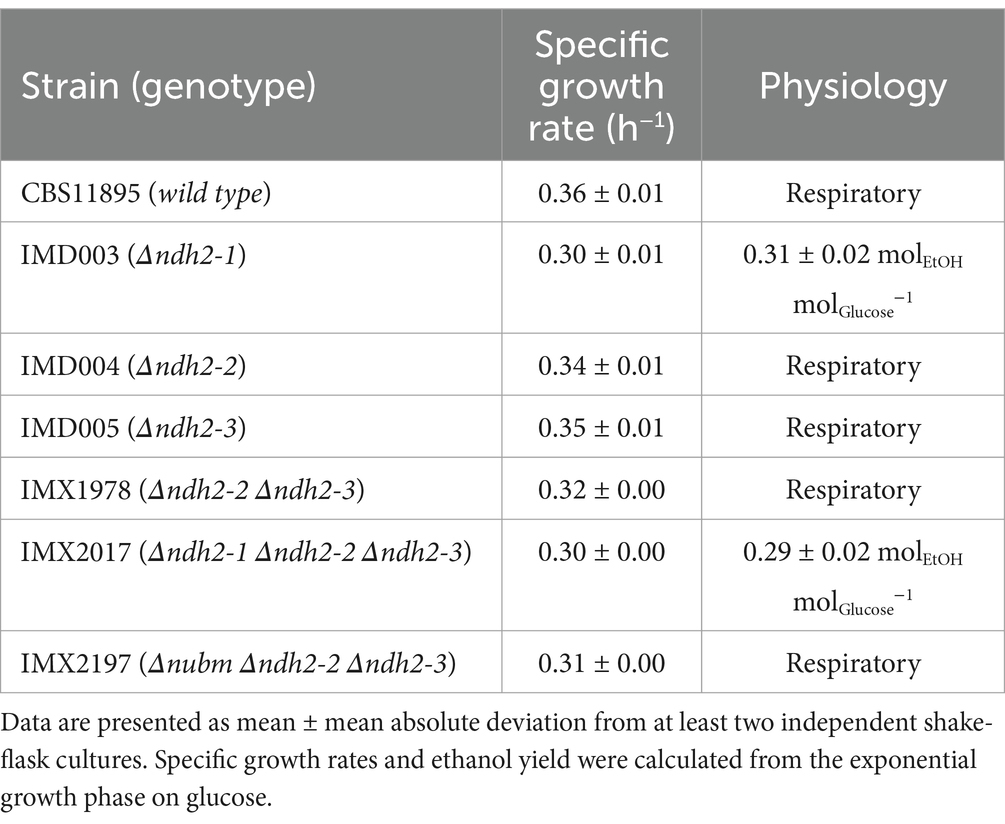
Table 1. Physiology of wild-type O. parapolymorpha CBS11895 and congenic mutant strains in aerobic shake-flask cultures grown at 30°C in synthetic medium with urea as nitrogen- and glucose (2 g L−1) as carbon source.
In shake-flask cultures, the specific growth rate of mutant strain IMD005 (Δndh2-3) did not differ significantly from that of the wild-type strain CBS11895, whereas strains IMD003 (Δndh2-1) and IMD004 (Δndh2-2) exhibited 17 and 6% lower specific growth rates, respectively (Table 1). In cultures of strains CBS11895, IMD004 and IMD005 no fermentation products were detected, indicating that a disruption of either NDH2-2 or NDH2-3 did not impede aerobic respiratory metabolism. In contrast, strain IMD003 exhibited a Crabtree-positive phenotype, producing 0.31 ± 0.02 mol of ethanol for each mol of glucose consumed.
We then combined disruptions in NDH2-2 and NDH2-3, resulting in strain IMX1978, which exhibited a slightly lower specific growth rate than the strains with individually disrupted NDH genes, but still maintained respiratory metabolism (Table 1). An additional disruption in strain IMX1978 of NUBM, which encodes an essential 51 kDA subunit of Ndh1 (Fecke et al., 1994), also did not result in detectable ethanol production in the resulting strain IMX2197 (Δnubm(ndh1) Δndh2-2 Δndh2-3). Collectively, these results demonstrate that Ndh2-1 alone is capable of supporting NAD(P)H turnover requirements in O. parapolymorpha for fully respiratory growth in a glucose grown batch culture.
Oxygen consumption studies with mitochondria from wild-type Ogataea parapolymorpha and NDH-2 deletion mutants confirm internal orientation of Ndh2-1Strain IMX2017, in which all three NDH2 genes were disrupted, exhibited a phenotype similar to that of strain IMD003 (Δndh2-1), displaying a 17% lower specific growth rate than observed for the wild-type strain CBS11895, and an ethanol yield similar to IMD003 (Table 1). Since yeast respiratory chains typically possess either none, or a single internal alternative NAD(P)H dehydrogenase (Antos-Krzeminska and Jarmuszkiewicz, 2019), and respiratory Complex I is not physiologically relevant under these conditions in O. parapolymorpha (Juergens et al., 2020), these data support the hypothesis that Ndh2-1 has an internal orientation (i.e., facing the mitochondrial matrix).
Oxygen-uptake rates of isolated mitochondria using a compartmentalized substrate approach has been previously used to determine the orientation of yeast mitochondrial NAD(P)H dehydrogenases (Luttik et al., 1998; Tarrio et al., 2005; Gonzalez-Barroso et al., 2006). To obtain O. parapolymorpha mitochondria, we adapted a protocol for isolation of mitochondria from glucose-limited S. cerevisiae cultures (Luttik et al., 1998). Initial isolations from wild-type cells grown in glucose-limited chemostat cultures resulted in well-coupled O. parapolymorpha mitochondria with a ‘respiratory control ratio’ (RCR) of 3.6 when assayed with endogenous NADH (generated in the mitochondrial matrix by addition of pyruvate and malate). However, the same preparations exhibited rapid, uncoupled oxygen uptake when exposed to methanol or ethanol. Methanol and ethanol dependent oxygen uptake rates were approximately 4-fold and 2.5-fold faster, respectively, than ADP-stimulated respiration of endogenous NADH. (M)ethanol-dependent oxygen uptake indicated a minor contamination of the mitochondrial preparations of these glucose-de-repressed cultures with peroxisomes containing methanol oxidase (MOX) (Egli et al., 1980; Yurimoto et al., 2011). A similar contamination was previously observed in mitochondrial preparations from the methylotrophic yeast Pichia pastoris (Gonzalez-Barroso et al., 2006). In an attempt to obtain mitochondrial preparations devoid of MOX activity, mitochondria were also isolated from cells grown under MOX-repressing conditions in glucose-grown batch- or nitrogen-limited chemostat cultures (Egli and Quayle, 1986; Ravin et al., 2013). Mitochondrial isolations from cells grown under these conditions did not consume oxygen in the presence of methanol, but they exhibited RCRs close to 1 when assayed with endogenous NADH, indicating uncoupled preparations (data not shown). In light of these findings, mitochondria isolated from glucose-limited chemostat cultures were used for respiration studies, and interference of the oxygen-uptake measurements by MOX activity was minimized by using reaction mixtures and substrates devoid of alcoholic solvents (see Methods section).
To test the hypothesis that the NADH binding site of Ndh2-1 is oriented toward the mitochondrial matrix, oxygen uptake was measured in the presence of endogenous and exogenous NADH using mitochondria isolated from wild type O. parapolymorpha (CBS11895), from strains possessing only Ndh1 (IMX2017; Δndh2-1 Δndh2-2 Δndh2-3) or only Ndh2-1 (IMX2197; Δnubm; Δndh2-2; Δndh2-3), henceforth known as ‘respiration-linked NAD(P)H dehydrogenases’ (Table 2). These strains were of interest because in aerobic glucose-limited chemostat cultures at a dilution rate of 0.1 h−1, all three strains exhibited a fully respiratory metabolism (see below, Table 3), indicating a functional respiratory chain capable of (re)oxidation of both cytosolic and mitochondrial NADH pools.
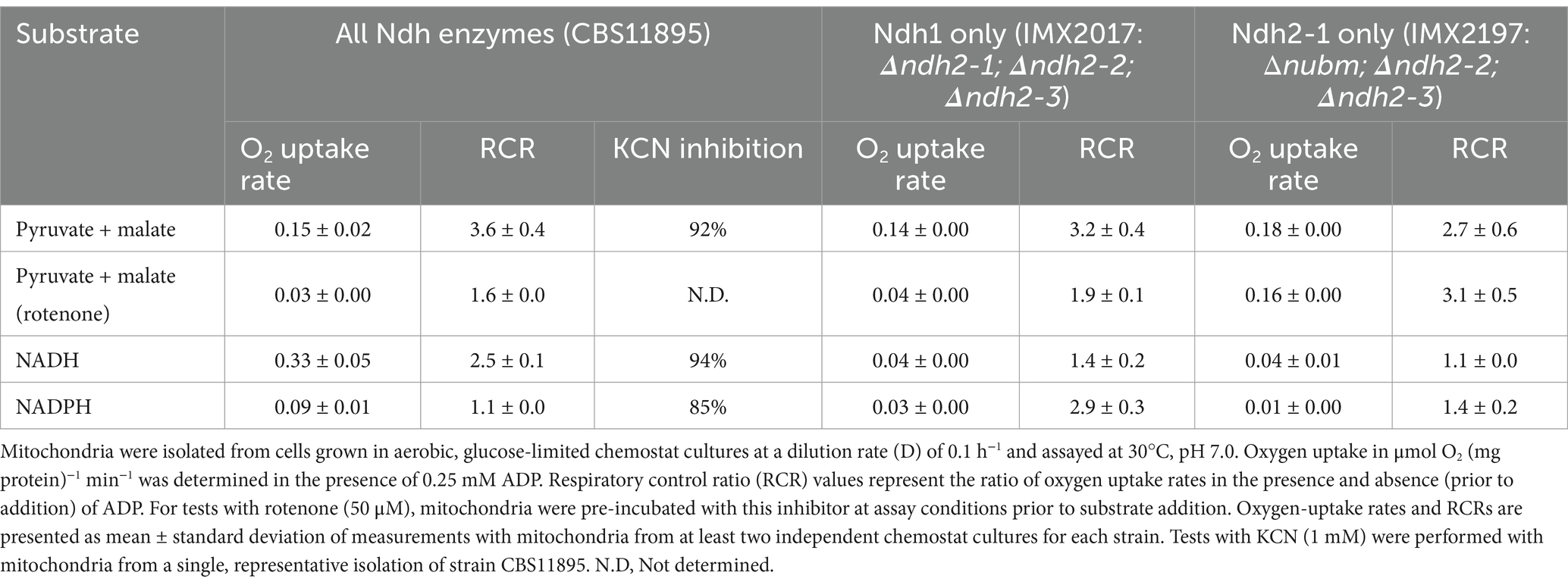
Table 2. Substrate-dependent rates of oxygen uptake by mitochondria from wild-type O. parapolymorpha (CBS11895) and mutants possessing only Ndh1 (IMX2017) or Ndh2-1 (IMX2197) as known respiration-linked NAD(P)H dehydrogenases.

Table 3. Physiology of Ogataea parapolymorpha strains CBS11895 (wild type), IMX2017 (Ndh1 only) and IMX2197 (Ndh2-1 only) in aerobic, glucose-limited chemostat cultures grown at a dilution of 0.1 h−1, 30°C and pH 5.
Mitochondria isolated from wild type O. parapolymorpha (CBS11895) readily consumed oxygen in the presence of endogenous and exogenous NADH (Table 2). RCRs for endogenous NADH (3.6 ± 0.4) and exogenous NADH (2.5 ± 0.1) indicated that the observed respiration linked NADH oxidation is due to the activity of separate internal and external NADH dehydrogenases, and not the result of physically compromised mitochondria. Furthermore, the near-complete inhibition of oxygen utilization by the cytochrome c oxidase inhibitor cyanide (~92 and ~94% effective in the presence of endogenous and exogenous NADH, respectively) strongly suggests that oxygen consumption was dependent on an intact oxidative membrane-bound electron transport chain capable of maintaining a proton motive force sufficient for ATP synthesis. In contrast, mitochondria from strains IMX2017 (Ndh1 only) and IMX2197 (Ndh2-1 only) both exhibited 88% lower oxygen consumption rates in the presence of exogenous NADH, suggesting that mitochondria from these strains do not possess any notable external NADH dehydrogenase activity characteristic of a type-II NADH dehydrogenase.
Mitochondria from strains IMX2017 (Ndh1 only) and IMX2197 (Ndh2-1 only) when assayed with endogenous NADH exhibited similar coupled oxygen-uptake rates to mitochondria isolated from the wild-type strain CBS11895 (Table 2). Presence of rotenone, the Ndh1-specific inhibitor, strongly decreased oxygen-uptake rates with endogenous NADH of mitochondria from the strains CBS11895 and IMX2017 (by 80 and 71%, respectively), while for strain IMX2197 no significant rotenone inhibition was observed. These observations demonstrate that Ndh2-1 is indeed internally oriented and able to completely take over the role of Ndh1 in NADH oxidation in Δnubm mutants under aerobic, glucose-limited conditions.
When mitochondria isolated from the wild-type strain O. parapolymorpha CBS11895 were assayed with exogenous NADPH, oxygen consumption was detected at a rate of 0.09 ± 0.01 μmol O2 (mg protein)−1 min−1, substantially lower than the rate obtained with exogenous NADH (0.33 ± 0.05 μmol O2 (mg protein)−1 min−1, Table 2). NADPH-dependent oxygen consumption was not significantly coupled (RCR of 1.1). However, it was largely inhibited by cyanide (~85%), indicating that NADPH oxidation did not occur via a soluble enzyme but by an external NADPH-accepting dehydrogenase that transferred electrons from NADPH into the mitochondrial respiratory chain. In mitochondria isolated from strains IMX2017 and IMX2197 and challenged with cyanide, exogenous NADPH oxidation activity was reduced by 3- and 9-fold, respectively, strongly suggesting that Ndh2-2 and/or Ndh2-3 is responsible for the NADPH oxidation observed in CBS11895 mitochondria.
Ogataea parapolymorpha mutants with linearized respiratory chains exhibit respiratory physiology in glucose-limited chemostat culturesIn aerobic glucose-limited chemostat cultures grown at a dilution rate of 0.1 h−1, both strain IMX2017 (Ndh1 only; Δndh2-1 Δndh2-2 Δndh2-3) and strain IMX2197 (Ndh2-1 only; Δnubm Δndh2-2 Δndh2-3) exhibited essentially the same respiratory phenotype as the wild-type strain CBS11895. Despite the deletion of one respiration-linked NAD(P)H dehydrogenase, both mutant strains grew without detectable formation of fermentation products. Moreover, an oxidative respiratory quotient close to 1 was observed and the carbon contained in the glucose feed could be completely recovered as biomass and carbon dioxide (Table 3). IMX2017 exhibited a biomass yield of 0.52 g biomass (g glucose)−1, which is not significantly different from that found with wild type CBS11895 (0.51 g biomass (g glucose)−1). In contrast, the biomass yield of strain IMX2197 was reduced by ~15% compared to that of the two other strains. This reduced biomass yield is consistent with a less efficient oxidative respiratory chain, using internal, non-proton pumping Ndh2-1 instead of the proton-pumping Ndh1. Accordingly, IMX2197 exhibited an approximately ~30% lower biomass yield on oxygen, with correspondingly higher biomass-specific rates of oxygen consumption and carbon-dioxide production.
The Ogataea parapolymorpha NDH2s oxidize NADH but not NADPH when expressed in Escherichia coli membranesThe oxygen-consumption experiments indicated that mitochondria from O. parapolymorpha poorly couple oxidation of exogenous NADPH to oxygen consumption via the aerobic respiratory chain. In principle, any external Ndh-2 could be responsible for this activity. Upon closer examination of the putative amino acid sequence of O. parapolymorpha Ndh2-3, an uncharged residue (Q365) instead of a negatively charged (E272 in S. cerevisiae Ndi1) is present within the substrate-binding domain (Supplementary Figure S2). This residue has been suggested to be involved in determining NADH/NADPH specificity due to interaction with the phosphate group of NADPH (Iwata et al., 2012; Michalecka et al., 2004).
To assess the ability of the O. parapolymorpha Ndh-2 to catalyze the oxidation of NADH and/or NADPH, NDH2-1, NDH2-2 and NDH2-3 were individually overexpressed in Escherichia coli, following a strategy previously applied to an NDH-2 (NDI1) from S. cerevisiae (Kitajima-Ihara and Yagi, 1998) and Caldalkalibacillus thermarum (Godoy-Hernandez et al., 2019; Godoy-Hernandez and McMillan, 2021). Since respiration-linked NADPH-dehydrogenase activity has not been reported for E. coli, this host was regarded as especially suitable to assess NADPH-dehydrogenase activity of heterologously expressed genes.
In spectrophotometric assays at pH 7.4, expression of each of the three O. parapolymorpha NDH-2 led to 2.4–3.2-fold higher NADH-oxidation rates than observed with membranes isolated from the parental E. coli strain (Figure 1). This activity indicated successful overexpression and localization to the E. coli membrane, and confirmed that the three O. parapolymorpha Ndh-2 are indeed NADH:quinone oxidoreductases. When NADPH was added as substrate to the same membrane preparations, no detectable NADPH-oxidation was measurable, indicating that none of the three O. parapolymorpha Ndh-2 can effectively utilize NADPH under the conditions tested (Supplementary Table S1). Since NAD(P)H oxidation by Ndh-2 can depend on pH (Melo et al., 2001; Nantapong et al., 2005), NADH and NADPH oxidation measurements were repeated at pH 5.5 and 8.0. These different pH values did not influence NADH oxidation rates relative to rates measured at pH 7.4 (Student’s t test, p > 0.05), and did not stimulate NADPH utilization by either endogenous E. coli respiratory enzymes or O. parapolymorpha Ndh-2 (Supplementary Table S1). Finally, oxidative NAD(P)H catalysis by Ndh2-3 overexpressed in E. coli was also tested in the presence of calcium, as Ndh2-3 contains a putative EF-hand suggesting a calcium binding domain which could potentially regulate catalytic behavior (Supplementary Figure S3; Melo et al., 2001). However, the presence of 5 mM Ca2+ did not significantly affect rates of NADH oxidation by Ndh2-3 overexpression membranes at pH 5.5 and 7.4 (24% reduction at pH 8.0) and did not enable NADPH utilization at pH 5.5, 7.4 or 8.0 (Supplementary Table S1).
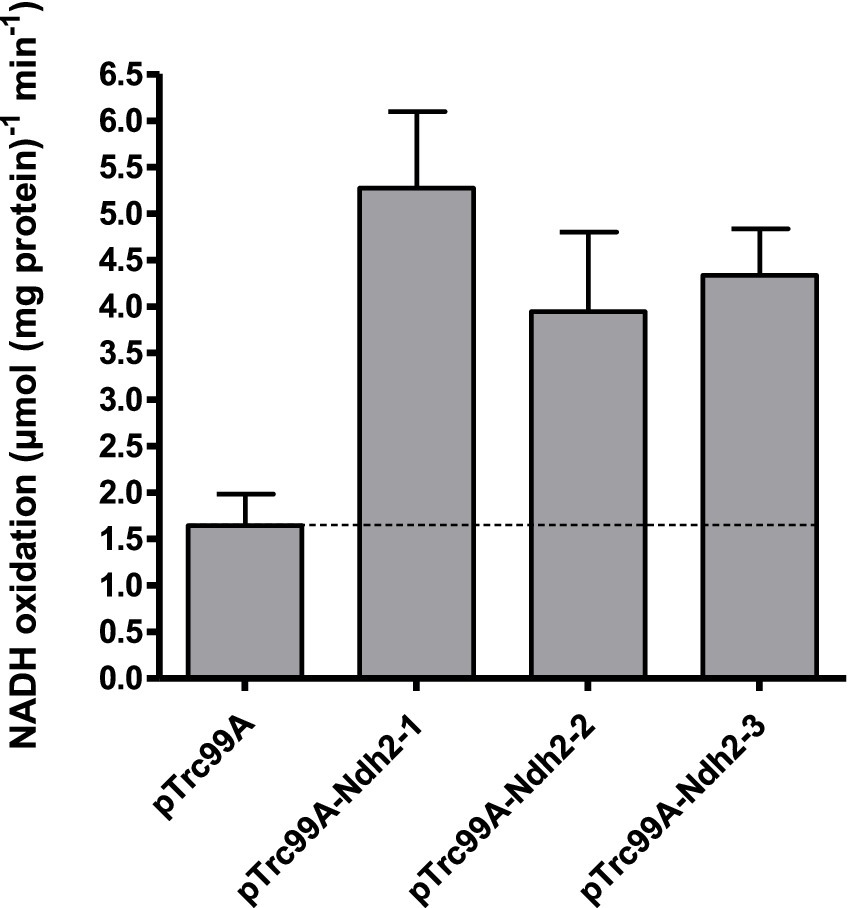
Figure 1. NADH oxidation by E. coli membranes isolated from strains overexpressing individual O. parapolymorpha NDH-2. Control measurements were done with membranes isolated from a strain carrying an empty overexpression plasmid (pTrc99A). Assays were performed with a membrane protein concentration of 10 μg mL−1 at 37°C, with 200 μM NADH and 100 μM ubiquinone-1. Data is presented as mean ± standard deviation of triplicate measurements.
Taken together, these data conclusively show that when assessed in E. coli membranes, the three NDH2s from O. parapolymorpha can efficiently oxidize NADH at a range of pH levels (pH 5.5–8.0), but do not catalyze the oxidation of NADPH.
Discussion NAD(P)H dehydrogenases in the respiratory chain of Ogataea parapolymorphaIn this study, the O. parapolymorpha gene HPODL_02792 (NDH2-1) was demonstrated to encode a mitochondrial ‘internal’ alternative NADH dehydrogenase. To achieve consistent nomenclature with other fungal species, we suggest naming this gene OpNDI1 (NADH dehydrogenase, internal). The closely related yeast O. polymorpha likely also possesses an internal alternative NADH dehydrogenase, as its genome (Riley et al., 2016) encodes a protein (OGAPODRAFT_15258) that exhibits 98% sequence identity with OpNdi1.
Mitochondria isolated from O. parapolymorpha also oxidized externally supplied NADH, indicating that either one, or both of the remaining alternative dehydrogenases, Ndh2-2 and Ndh2-3, are facing the intermembrane space, rather than the mitochondrial matrix. Coexistence of more than one internal alternative dehydrogenase has been described (Rasmusson et al., 2004), but since the fungi and yeasts so far characterized either possess one or no such enzymes, both Ndh2-2 and Ndh2-3 appear to be externally oriented. The internal orientation of Ndh2-1/OpNdi1 has now been established in this study.
Wild-type O. parapolymorpha mitochondria also oxidized NADPH via the respiratory chain as demonstrated by near-complete inhibition of this activity by cyanide. While growing on glucose the production of NADPH can be balanced with biosynthetic requirements, assimilation of other carbon sources such as gluconate result in a surplus of NADPH in yeast (Bruinenberg et al., 1983a), making the ability to respire NADPH a beneficial trait. The oxygen-uptake activity with NADPH was essentially uncoupled (RCR of 1.1), which was unexpected, as addition of ADP should relieve the backpressure of the proton gradient, especially since the same mitochondrial preparations exhibited well-coupled oxygen uptake in the presence of NADH. Mitochondria isolated from glucose-limited chemostat cultures of K. lactis (D = 0.1 h−1) similarly exhibited uncoupled oxygen uptake in the presence of NADPH (Overkamp et al., 2002), while mitochondria from lactate- and glucose-grown batch cultures of the same yeast exhibited RCRs of 2.3 and 1.3 for NADPH, respectively (Tarrio et al., 2006). With mitochondria isolated from glucose-limited chemostats of Candida utilis, the RCRs of NADPH oxidation were found to vary as a function of the dilution rate, with cultures grown at D = 0.05–0.1 h−1 exhibiting an RCR of ~1.9 while cultures grown at D = 0.2–0.4 h−1 exhibited a lower RCR of ~1.2 (Bruinenberg et al., 1985a). In agreement with our study, these studies reported inhibition of NADPH oxidation by cyanide and/or antimycin A, indicating that the degree of oxidative coupling of NADPH respiration by yeast mitochondria is dependent on the utilized substrate and/or the growth condition employed, which might also be the case in O. parapolymorpha.
The heterologous expression of O. parapolymorpha NDH-2 in E. coli showed that NADPH oxidation of the O. parapolymorpha mitochondria is not likely to have occurred via these Ndh-2, since all three O. parapolymorpha Ndh-2 exclusively utilized NADH when assayed within E. coli membranes. Based on their protein sequences, we originally speculated Ndh2-3 to be the most likely candidate to accept NADPH, as it contains the uncharged residue (Q365) proposed to permit NADPH utilization (Michalecka et al., 2004). There are indeed NADPH-utilizing NDH2s with this uncharged residue such as Nde1 from N. crassa (Supplementary Figure S1) or plant enzyme St-NBD1 (Michalecka et al., 2004), and mutation of this exact residue has been exploited to alter substrate specificity from NADH to co-utilization of NADH and NADPH in a bacterial NDH2 (Desplats et al., 2007). However, Nde2 from N. crassa and Nde1 from K. lactis do not contain the uncharged residue but still accept both substrates (Supplementary Figure S2), indicating that a charged amino acid in this position does not strictly prevent NADPH utilization and that Ndh-2 substrate specificity cannot be accurately predicted by a single residue. Another possibility is that Ndh2-3 could be post-translationally modified in O. parapolymorpha, in this instance, a post-translational modification (PTM) could theoretically tune the activity of Ndh2-3 toward NADPH. However, this possibility is unlikely and no enzyme has been reported to date to have a PTM to modify active site substrate specificity, moreover, we could also not detect any yeast PTM sites near the nucleotide binding sites using the YAAM database (Ledesma et al., 2018). In contrast to the NADPH utilizing Nde1 from N. crassa, where NADPH oxidation is highly affected by calcium (Melo et al., 2001), the putative calcium binding domains of Ndh2-3 from O. parapolymorpha and Nde2 from K. lactis are poorly conserved, and apparently lost their regulatory function, as presence of calcium did not appear to have an effect on activity of either enzyme (Tarrio et al., 2006). Collectively, this supports the notion that the three type-II Ndh from O. parapolymorpha are unlikely to be responsible for the observed NADPH oxidation activity, indicating the presence of another unidentified enzyme.
Mechanisms independent of Ndh-2 for turnover of cytosolic NADHMitochondria from strains IMX2017 (Ndh1 only; Δndh2-1 Δndh2-2 Δndh2-3) and IMX2197 (Ndh2-1 only; Δnubm Δndh2-2 Δndh2-3) did not oxidize external NADH, but both strains exhibited a fully respiratory phenotype in glucose-limited chemostat cultures at D = 0.1 h−1. Since both strains only contain a NADH dehydrogenase that can accept electrons from NADH in the mitochondrial matrix (Ndh1 and Ndh2-1/OpNdi1, respectively), mechanisms other than the external alternative NADH dehydrogenases are capable of respiring cytosolic NADH in these strains must be present. One candidate would be the ‘Gut2/Gpd shuttle’, consisting of mitochondrial glycerol-3-phosphate dehydrogenase (Gut2), which indirectly respires cytosolic NADH in combination with cytosolic NAD-dependent glycerol 3-phosphate dehydrogenase (Gpd) (Bakker et al., 2001). When grown on glycerol, O. parapolymorpha exhibits highly increased glycerol-kinase activity, indicating that glycerol assimilation occurs via the phosphorylative pathway. This suggests that Gut2 is functional in this yeast (Tani and Yamada, 1987). Furthermore, Gut2 activity was demonstrated in the closely related yeast O. polymorpha (de Koning et al., 1987). In S. cerevisiae, the metabolic function of Gut2 overlaps with the external Ndh-2 (Larsson et al., 1998; Overkamp et al., 2000), and yeast mitochondria isolated from glucose-grown cultures of various yeast species have been demonstrated to respire glycerol-3-phosphate with similar specific oxygen uptake rates as NADH (Luttik et al., 1998; Gonzalez-Barroso et al., 2006; Overkamp et al., 2002).
While S. cerevisiae lacking Gut2 in addition to its two external NADH dehydrogenases produces large amounts of glycerol under aerobic, glucose-limited conditions (Palmieri et al., 2006), an O. parapolymorpha mutant lacking Gut2 in addition to the three Ndh-2 (Δndh2-1 Δndh2-2 Δndh2-3 Δgut2) exhibited fully respiratory physiology under identical conditions (Supplementary Table S2). The absence of byproduct formation indicates that at least one additional, unknown mechanism for coupling the oxidation of cytosolic NADH to mitochondrial respiration is present in O. parapolymorpha. Such a mechanism could, for example, involve a shuttle for import of NADH equivalents into the mitochondrial matrix, where oxidation by Complex I and/or OpNdi1 can occur. For S. cerevisiae, several suitable mechanisms have been demonstrated or proposed, such as the malate-oxaloacetate and malate–aspartate shuttles, mitochondrial oxidation of ethanol produced in the cytosol, or, assuming a high cytosolic NADH/NAD+ ratio, an inward-directed ethanol-acetaldehyde shuttle (Bakker et al., 2001; Palmieri et al., 2006). Based on sequence homology with S. cerevisiae, key proteins for these mechanisms are present in O. parapolymorpha (Supplementary Table S3), however their operation and physiological relevance has not been confirmed by functional analysis studies.
Physiological relevance of branched respiratory chainsRespiratory chains of plants, fungi and some protists are branched (Antos-Krzeminska and Jarmuszkiewicz, 2019; Rasmusson et al., 2008), and, as also demonstrated in this study for O. parapolymorpha, can exhibit a metabolic redundancy for respiration-linked NADH oxidation, minimizing the physiological effect of loss of individual or multiple NADH dehydrogenases (Carneiro et al., 2004; Overkamp et al., 2000; Fromm et al., 2016). In general, this redundancy and the exact physiological role of the alternative NADH dehydrogenases are still poorly understood. In O. parapolymorpha, OpNdi1 appears to have a unique metabolic function as it is the only NADH dehydrogenase strictly required to sustain fast respiratory growth under glucose excess conditions, demonstrating that limiting respiratory capacity by disruption of a single NADH dehydrogenase can elicit the Crabtree effect in a Crabtree-negative yeast. Similarly, the overexpression of a single ‘Gal4-like’ transcription factor has been reported to convert Crabtree-negative P. pastoris into a Crabtree-positive yeast (Ata et al., 2018). However, since formation of ethanol and CO2 from glucose by alcoholic fermentation is redox-neutral and reduced by-products such as glycerol were not detected in fermenting cultures of IMD003 (Δndh2-1) and IMX2017 (Δndh2-1 Δndh2-2 Δndh2-3), (re)oxidation of mitochondrial NADH from oxidative sugar metabolism must still occur via respiration. While Ndh1 has been demonstrated to be physiologically irrelevant under these glucose excess conditions in wild-type O. parapolymorpha CBS11895 (Juergens et al., 2020), based on the available data its contribution to oxidation of mitochondrial NADH cannot be excluded in these strains. Alternatively, if export of NADH equivalents from the mitochondrial matrix is possible in this yeast, NADH could also be oxidized in the cytosol, for example by external NADH dehydrogenase(s) in IMD003 (Δndh2-1) or the Gut2/Gpd shuttle in IMX2017 (Ndh1 only).
A previous study with sub-mitochondrial particles harvested from stationary-phase cultures of O. polymorpha found that most NADH oxidation occurred via Ndh-2 and only ~10% via Ndh1 (Bridges et al., 2009). Similarly, in our experiments with mitochondria isolated from wild-type O. parapolymorpha grown in glucose-limited chemostat cultures, only ~30% of the total specific NADH oxidation activity could be attributed to Ndh1. However, due to the saturating substrate concentrations used in these types of experiments, they allow only for a limited interpretation of the actual physiological relevance of the respective enzymes. In vivo, competition of Complex I and OpNdi1 for NADH likely occurs if both systems are expressed at the same time, and indeed some fungal species appear to co-utilize Complex I and internal Ndh-2 (Fecke et al., 1994; Prömper et al., 1993; Voulgaris et al., 2012). With mitochondria isolated from wild-type O. parapolymorpha strain CBS11895, the presence of 50 μM rotenone could not fully abolish internal NADH oxidation. However, as strain IMX2017 (Ndh1 only), exhibited a similar partial inhibition of oxygen uptake by rotenone, the uninhibited activity in CBS11895 was likely not caused by OpNdi1 but instead by incomplete inhibition of O. parapolymorpha Ndh1. Comparative studies with sub-mitochondrial particles have demonstrated that rotenone inhibits Ndh1 from yeasts less strongly than the Bos taurus enzyme, requiring 50 μM rotenone to achieve 96% inhibition of NADH oxidation activity by the P. pastoris Ndh1 (Bridges et al., 2009). It is conceivable that Ndh1 from O. parapolymorpha is even more resistant to rotenone, explaining the observed partial inhibition in strains CBS11895 (wild type) and IMX2017 (Ndh1 only). In addition, OpNdi1 was not detected in the proteome of aerobic, glucose-limited cultures of CBS11895 (Juergens et al., 2020), and the observed identical biomass yields of strains CBS11895 and IMX2017 are consistent with a situation in which essentially all mitochondrial NADH is (re)oxidized by Ndh1 in wild-type O. parapolymorpha under these conditions. These observations indicate that oxidation of mitochondrial NADH is strictly separated between Ndh1 and OpNdi1 in O. parapolymorpha under conditions of glucose limitation and glucose excess, respectively. Nevertheless, OpNdi1 is able to fully support respiratory growth in the absence of a functional Complex I, as previously hypothesized (Juergens et al., 2020).
Concluding remarksIn this study we show that the Crabtree-negative yeast O. parapolymorpha contains both NDH1- and NDH2-type NADH dehydrogenases for respiration of NADH in the mitochondrial matrix but limits their utilization to conditions of carbon limitation and carbon excess, respectively. Furthermore, we find that the respiratory chain of O. parapolymorpha can tolerate multiple deletions without compromising respiratory metabolism, offering insight into its flexible nature and opportunities for metabolic (redox) engineering, for example to increase cytosolic NADH availability, in this industrially relevant yeast. Finally, the phenotype elicited by disruption of OpNdi1 demonstrates that limiting respiratory capacity by a single mutation in an NADH dehydrogenase can result in overflow metabolism and convert O. parapolymorpha into a yeast with a Crabtree-positive phenotype.
Materials and methods Yeast strains and maintenanceThe O. parapolymorpha strains used in this study are derived from the wild-type strain CBS11895 (DL-1; ATCC26012) and are described in Table 4. For construction and maintenance, yeast strains were grown in an Innova shaker incubator (New Brunswick Scientific, Edison, NJ, USA) set to 30°C and 200 rpm, in 500 mL shake flasks containing 100 mL heat-sterilized (120°C for 20 min) YPD medium (10 g L−1 Bacto yeast extract, 20 g L−1 Bacto peptone, 20 g L−1 glucose, demineralized water). Solid medium was prepared by addition of 2% (w/v) agar. Frozen stock cultures were prepared from exponentially growing shake-flask cultures by addition of glycerol to a final concentration of 30% (v/v), and aseptically stored in 1 mL aliquots at −80°C.
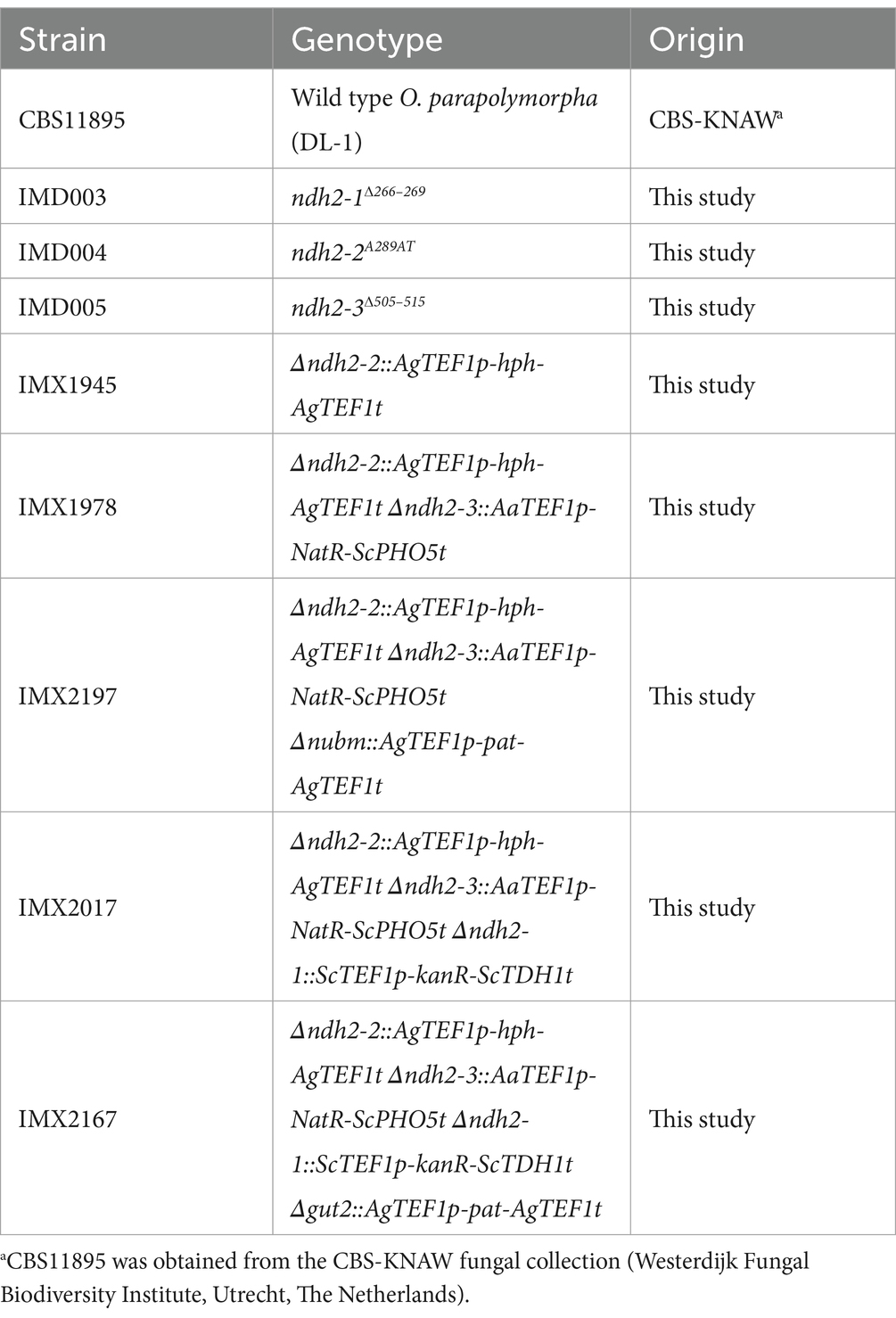
Table 4. O. parapolymorpha (Hansenula polymorpha) strains used in this study.
Plasmid constructionAll plasmids used in this study are described in Table 5. Plasmids pUD546, pUD547 and pUD548 were de novo synthesized by GeneArt (Thermo Fisher Scientific, Waltham, MA, USA) and contained synthetic guide RNA (gRNA) constructs with spacer sequences (5′-3′) ‘CCTGATGTAAATATACGCTG’, ‘AAGAAGAACATTGTTATTCT’, and ‘GTTATTCTGGGTTCCGGCTG’, respectively. Cas9/gRNA co-expression plasmids pUDP019, pUDP020 and pUDP021 were constructed using pUD546, pUD547 and pUD548, respectively, by integration into pUDP002 via BsaI-mediated ‘Golden Gate’ assembly (Engler et al., 2008) as described previously (Juergens et al., 2018), and verified by digestion with PdmI.
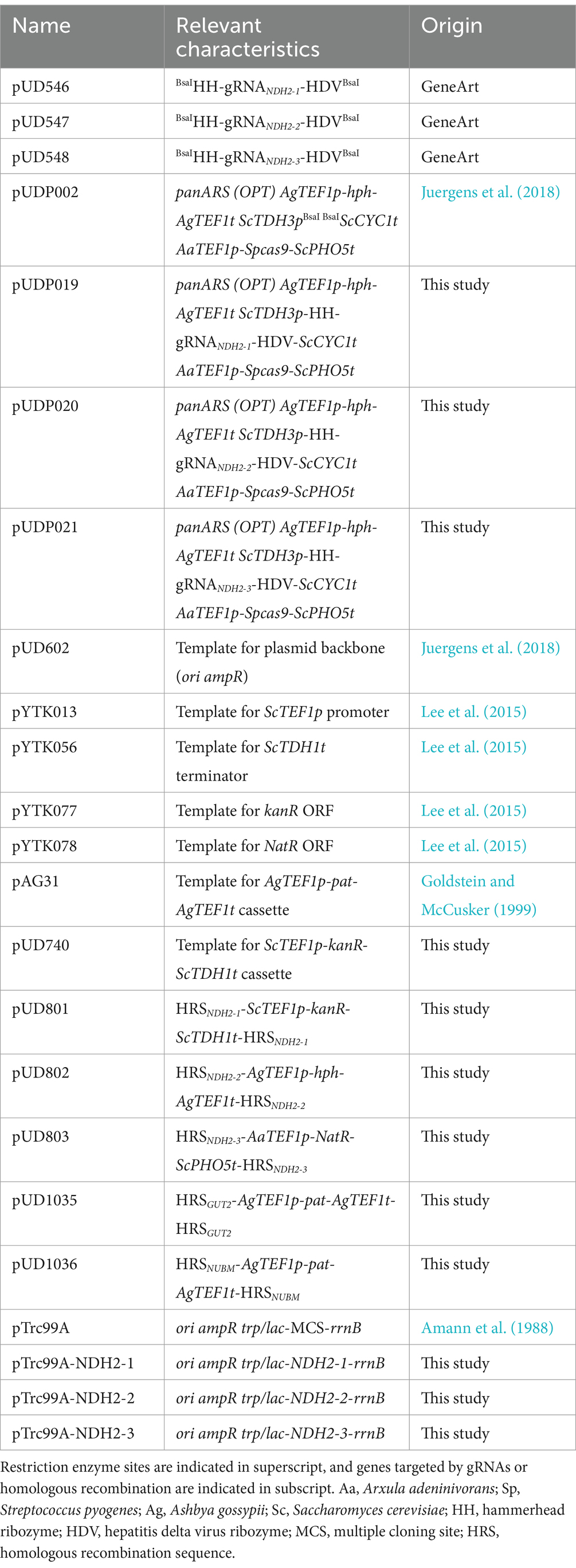
Table 5. Plasmids used in this study.
Construction of pUD740 was done via ‘Gibson assembly’ (Gibson et al., 2009) from the following PCR-amplified fragments: the ScTEF1 promoter from pYTK013 (primers 12099 + 12100), the kanR G418 resistance marker ORF from pYTK077 (primers 12097 + 12098), the ScTDH1 terminator from pYTK056 (primers 12095 + 12096), the AaTEF1p-Spcas9-ScPHO5t expression cassette from pUDP002 (primers 10426 + 10427), upstream (primers 12093 + 12094) and downstream (primers 12101 + 12102) homologous recombination sequences of the OpKU70 locus from CBS11895 genomic DNA and the plasmid backbone from pUD602 (primers 12103 + 12104).
Plasmids pUD801, pUD802, pUD803, pUD1035 and pUD1036 carrying subcloned homology-flanked marker cassettes for split-marker deletion were constructed by Gibson assembly from various PCR-amplified fragments. For the construction of pUD801 these fragments were: the ScTEF1p-kanR-ScTDH1t G418 resistance marker cassette from pUD740 (primers 12100 + 4377), upstream (primers 14385 + 14386) and downstream (primers 14387 + 14388) homologous recombination sequences of the NDH2-1 locus from CBS11895 genomic DNA and the plasmid backbone from pUD602 (primers 12103 + 12104). For pUD802 these fragments were: the AgTEF1p-hph-AgTEF1t hygromycin resistance marker cassette from pUDP002 (primers 11065 + 11133), upstream (primers 14389 + 14390) and downstream (primers 14391 + 14392) homologous recombination sequences of the NDH2-2 locus from CBS11895 genomic DNA and the plasmid backbone from pUD602 (primers 12103 + 12104). For pUD803 these fragments were: the AaTEF1 promoter from pUDP002 (primers 10426 + 14383), the NatR nourseothricin resistance marker ORF from pYTK078 (primers 14381 + 14382), the ScPHO5 terminator from pUDP002 (primers 10427 + 14384), upstream (primers 14393 + 14394) and downstream (primers 14395 + 14396) homologous recombination sequences of the NDH2-3 locus from CBS11895 genomic DNA and the plasmid backbone from pUD602 (primers 12103 + 12104). For pUD1035 these fragments were: the AgTEF1p-pat-AgTEF1t phosphinothricin resistance marker cassette from pAG31 (primers 3242 + 8439), upstream (primers 14803 + 14804) and downstream (primers 14805 + 14806) homologous recombination sequences for GUT2 from CBS11895 genomic DNA and the plasmid backbone from pUD602 (primers 12103 + 12104). For pUD1036 these fragments were: the AgTEF1p-pat-AgTEF1t phosphinothricin resistance marker cassette from pAG31 (primers 3242 + 8439), upstream (primers 14807 + 14808) and downstream (primers 14809 + 14810) homologous recombination sequences for NUBM from CBS11895 genomic DNA and the plasmid backbone from pUD602 (primers 12103 + 12104). Correct insertion and presence of the homology-flanked marker cassettes in the constructed plasmids was verified by restriction digest and diagnostic PCR using PvuI + NdeI and primer sets 2908 + 12616 and 1642 + 3983 (pUD801), PvuI + PsiI and primer sets 2457 + 12616 and 1642 + 1781 (pUD802), PvuI + KpnI and primer sets 10459 + 12616 and 1642 + 10458 (pUD803) and PvuI + NcoI and primer sets 1409 + 12616 and 1642 + 4662 (pUD1035 & pUD1036).
Plasmids pTrc99A-NDH2-1, pTrc99A-NDH2-2 and pTrc99A-NDH2-3 were constructed by restriction/ligation cloning. The ORFs encoding NDH2-1 (HPODL_02792), NDH2-2 (HPODL_00256) and NDH2-3 (HPODL_02018) were PCR-amplified from CBS11895 genomic DNA using primer sets 14929 + 14931, 16075 + 16076 and 16077 + 16078, respectively. Amplification using these primer sets added a 5′ NcoI site, a GS-flanked 6-HIS tag (‘GSHHHHHHGS’) directly after the start codon and a 3′ XmaI site to all three ORFs. Furthermore, amplification of NDH2-1 omitted the first 24 amino acids after the start codon, as they could be unambiguously identified as mitochondrial targeting sequence by MitoFates (Fukasawa et al., 2015). PCR amplicons were then digested with NcoI and XmaI and cloned into NcoI/XmaI-digested pTrc99A using T4 DNA ligase (New England Biolabs, Ipswich, MA, USA).
Yeast strain constructionOgataea parapolymorpha strains were transformed via electroporation of freshly prepared electrocompetent cells as described previously (Juergens et al., 2018). Depending on the selection marker, mutants were selected on solid YPD medium supplemented with 200 μg mL−1 G418, 300 μg mL−1 hygromycin B or 100 μg mL−1 nourseothricin, or on solid synthetic medium (SM) supplemented with 20 g L−1 glucose and 200 μg mL−1 bialaphos (SanBio, Uden, The Netherlands). SM was prepared according to Verduyn, Postma (Verduyn et al., 1992) and autoclaved at 120°C for 20 min. Glucose and vitamins (Verduyn et al., 1992) were prepared separately and filter-sterilized (vitamins) or heat-sterilized at 110°C for 20 min (glucose).
Strains IMD003, IMD004 and IMD005 with disrupted versions of genes NDH2-1 (HPODL_02792), NDH2-2 (HPODL_00256) and NDH2-3 (HPODL_02018), respectively, were constructed using the pUDP CRISPR/Cas9 system described previously (Juergens et al., 2018). Wild type strain CBS11895 was transformed with pUDP019, pUDP020 or pUDP021 targeting NDH2-1 (after base part 269 out of 1,614), NDH2-2 (after base pair 290 out of 1,671) and NDH2-3 (after base pair 515 out of 2,097), respectively, and subjected to the prolonged liquid incubation protocol as described previously for deletion of OpADE2 and OpKU80 (Juergens et al., 2018). Randomly picked colonies were then subjected to PCR amplification of the NDH2-1, NDH2-2 and NDH2-3 locus using primer sets 10742 + 10743, 10744 + 10745 and 10746 + 10747, respectively, followed by Sanger sequencing (Baseclear, Leiden, The Netherlands) to identify mutant transformants harboring a frame-shifting indel at the respective gRNA target sites. Three mutants with either a deletion of base pairs 226–229 of NDH2-1, an additional thymine nucleotide between position 289 and 290 of NDH2-2 or a deletion of base pairs 505–515 of NDH2-3 were identified, restreaked three times subsequently on non-selective YPD medium to remove the pUDP plasmids, and renamed IMD003, IMD004 and IMD005, respectively.
Strains IMX1945, IMX1978, IMX2017, IMX2197 and IMX2167 were constructed using a split-marker deletion approach (Fairhead et al., 1996), with ~480 bp of internal (marker recombination) and ~480 bp of external (genome recombination) homology. To preserve the promoter and terminator sequences of neighboring genes and limit interference of their expression, a minimum of 800 bp preceding and 300 bp succeeding adjacent ORFs were kept unaffected by the deletions. IMX1945 was constructed from wild type O. parapolymorpha strain CBS11895 by deletion of NDH2-2 with a hyg resistance cassette using pUD802, IMX1978 was constructed from IMX1945 by additional deletion of NDH2-3 with a NatR resistance cassette using pUD803, IMX2017 was constructed from IMX1978 by additional deletion of NDH2-1 with a kanR resistance cassette using pUD801, IMX2197 was constructed from IMX1978 by additional deletion of NUBM (HPODL_04625) (Juergens et al., 2020) with a pat resistance cassette using pUD1036, and IMX2167 was constructed from IMX2017 by additional deletion of GUT2 (HPODL_00581) with a pat resistance cassette using pUD1035. For the split-marker deletion of NDH2-1, NDH2-2, NDH2-3, NUBM and GUT2, the two overlapping fragments for transformation were PCR-amplified from pUD801 using primer sets 6816 + 14397 and 12565 + 14398, from pUD802 using primer sets 14399 + 14400 and 14401 + 14402, from pUD803 using primer sets 14403 + 14404 and 14405 + 14406, from pUD1036 using primer sets 15884 + 15885 and 15886 + 15887, and from pUD1035 using primer sets 14811 + 15885 and 14812 + 15886, respectively. Prior to transformation, the amplified fragments were gel-purified and, in case DNA amounts were too low for transformation, used as template for another PCR amplification using the same primers followed by PCR purification. For each transformation, a total of ~1 μg purified DNA (both fragments equimolar) in a maximum volume of 4 μL was transformed to 40 μL of fresh electrocompetent cells as described above, with the exception that after electroporation the cell suspensions were recovered in 1 mL YPD for 3 h at 30°C before plating onto selective medium. Additionally, after YPD recovery, cells transformed with the pat resistance marker were washed once by centrifugation and resuspension in sterile demineralized water before selective plating. Selection plates were typically incubated for 3 days at 30°C before assessment of the correct replacement of the target genes with the resistance markers via diagnostic PCR using primer sets 14465 + 14466, 14465 + 4047 and 2653 + 14466 for Δndh2-1::kanR, primer sets 14467 + 14468, 14467 + 7864 and 8411 + 14468 for Δndh2-2::hph, primer sets 14469 + 14470, 14469 + 11197 and 11202 + 14470 for Δndh2-3::NatR, primer sets 15630 + 15631, 15630 + 15885 and 15886 + 15631 for Δnubm::pat and primer sets 15628 + 15629, 15628 + 15885 and 15886 + 15629 for ΔGUT2::pat. Single colonies that contained the desired genotype(s) were re-streaked once on selective medium, followed by two re-streaks on non-selective YPD medium before stocking.
Molecular biologyPCR amplification for cloning and construction was performed with Phusion High Fidelity Polymerase (Thermo Fisher Scientific) using PAGE-purified oligonucleotide primers (Sigma-Aldrich, St. Louis, MO, USA) according to manufacturer’s recommendations, with the exception that a final primer concentration of 0.2 μM was used. Diagnostic PCR was done using DreamTaq polymerase (Thermo Fisher Scientific) and desalted primers (Sigma-Aldrich). The primers used in this study are shown in Supplementary Table S4. Genomic DNA of yeast colonies was isolated using the LiAc-sodium dodecyl sulfate method (Lõoke et al., 2011) or the YeaStar Genomic DNA kit (Zymo Research, Irvine, CA, USA). DNA fragments obtained by PCR were separated by gel electrophoresis. Gel-purification was carried out using the Zymoclean Gel DNA Recovery Kit (Zymo Research). PCR purification was performed using the GenElute PCR Clean-Up Kit (Sigma-Aldrich). Gibson assembly was done using the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) with purified DNA fragments according to manufacturer’s recommendations, with the exception that reaction volume was down-scaled to 5–10 μL. DNA fragments that were PCR-amplified from a template harboring the same bacterial resistance marker as the construct to be Gibson-assembled were subjected to DpnI treatment prior to PCR cleanup. Restriction digest was performed using FastDigest enzymes (Thermo Fisher Scientific) or High Fidelity (HF) restriction endonucleases (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s instructions. Escherichia coli strains XL1-blue and DH5α were used for plasmid transformation, amplification and storage. Plasmid isolation from E. coli was done using the GenElute Plasmid Miniprep Kit (Sigma-Aldrich) or the Monarch Plasmid Miniprep Kit (New England Biolabs).
Multiple sequence alignment and domain predictionAlignment of Ndh-2 protein sequences was done using MUSCLE (Edgar, 2004) and visualized using Jalview (Waterhouse et al., 2009). Orientation and substrate specificity of other fungal Ndh-2 were taken from: K. lactis (Tarrio et al., 2005; Tarrio et al., 2006), N. crassa (Carneiro et al., 2004; Carneiro et al., 2007; Melo et al., 2001; Duarte et al., 2003), S. cerevisiae (de Vries and Marres, 1987; Luttik et al., 1998; Small and McAlister-Henn, 1998; Van Urk et al., 1989), and Y. lipolytica (Kerscher et al., 1999). Prediction of the putative EF-hand calcium binding domain in Ndh-2 sequences was done by Motif Scan using PROSITE profiles (Sigrist et al., 2010). Prediction of transmembrane helicies was performed using TMHMM—2.0 (Krogh et al., 2001).
Shake-flask cultivationShake-flask growth experiments were performed with synthetic medium with urea as nitrogen source to avoid fast acidification (Luttik et al., 2000), set to an initial pH of 5.0 with KOH. Cultures were grown in 500 mL round-bottom shake flasks filled with 50 mL medium. Cultures were grown with 2 g L−1 glucose as sole carbon source and were inoculated with mid-exponential precultures (washed once with sterile demineralized water) to an initial OD660 of 0.3. Precultures were grown under the same conditions and in the same medium, but with an initial glucose concentration of 5 g L−1. Shake flasks were continuously shaken during sampling to prevent oxygen limitation. Physiological parameters were calculated from at least five samples taken during the exponential growth phase. Calculated ethanol yields were not corrected for evaporation.
Chemostat cultivationChemostat cultivation was performed as described previously (Juergens et al., 2020) using SM with the addition of 0.15 g L−1 Pluronic 6100 PE antifoaming agent (BASF, Ludwigshafen, Germany) and glucose (7.5 or 9 g L−1) as sole carbon source. SM was prepared according to Verduyn, Postma (Lee et al., 2015) as described above. Bioreactors were inoculated with exponentially growing shake flask cultures (SM with 20 g L−1 glucose). Chemostat cultivation was performed in 2-L benchtop bioreactors (Applikon, Delft, The Netherlands) with a working volume of 1.0 L which was maintained by an electrical level sensor that controlled the effluent pump. The dilution rate was set by maintaining a constant medium inflow rate. Cultures were sparged with dried, compressed air (0.5 vvm) and stirred at 800 rpm. Temperature was maintained at 30°C and pH was controlled at 5.0 by automatic addition of a 2 M KOH by an EZcontroller (Applikon). The exhaust gas was cooled with a condenser (2°C) and dried with a Perma Pure Dryer (Inacom Instruments, Veenendaal, the Netherlands) prior to online analysis of carbon dioxide and oxygen with a Rosemount NGA 2000 Analyzer (Emerson, St. Louis, MO, USA). Cultures were assumed to have reached steady state when, after a minimum of 5 volume changes, the oxygen-consumption rate, carbon-dioxide production rate and biomass concentration changed by less than 3% over two consecutive volume changes.
Analytical methodsOptical density (OD) of yeast cultures was measured at 660 nm on a Jenway 7200 spectrophotometer (Jenway, Staffordshire, UK). OD of bacterial cultures was measured at 600 nm on Ultrospec 2100 pro (Amersham, Little Chalfont, UK). For biomass dry weight determination of yeast cultures, exactly 10 mL of culture broth was filtered over pre-dried and pre-weighed membrane filters
留言 (0)