
記住我
The severity of pertussis, an infectious respiratory disease caused by Bordetella pertussis (BP), is high in infants (Payne et al., 2023). Whole-cell pertussis vaccine (WCV) effectively reduced the occurrence of pertussis. In China, WCV was replaced by the less reactogenic acellular pertussis vaccine (ACV), which has been solely used in China since 2013 (Xu et al., 2015). ACV comprises pure BP proteins, especially filamentous hemagglutinin, pertussis toxin promoter (ptx) which is the principal pathogenic factor of BP, capable of inducing the immune system to produce antibodies, and pertactin (prn) which has robust immunogenicity and is involved in the adhesion mechanism among bacteria, neutrophils, and epithelial cells, with a few additional proteins, including fimbrial proteins (Belcher and Preston, 2015).
Although ACV is effective, the prevalence of BP has been increasing, which is a major global public health concern, especially in industrialized countries (Barkoff and He, 2019). The loss of immunity caused by ACV occurs earlier than that caused by WCV, increasing infection susceptibility in children and adolescents (Belcher and Preston, 2015).
One explanation for the increased circulation of BP is molecular alterations in the pathogen. Various studies have indicated antigenic changes in bacterial virulence genes that may suppress vaccine-mediated immunity against BP (Barkoff and He, 2019). Pertussis vaccination is reported to alter the fimbrial serotype of the circulating strains. Advances in bacterial typing techniques have enabled the elucidation of differential characteristics between vaccination and epidemic strains. In particular, alterations have been detected in the shared virulence genes, such as ptx and prn (Barkoff and He, 2019). Additionally, genome-based methods, such as multi-locus sequence typing (MLST), multi-locus antigen sequence typing (MAST), and whole-genome sequencing (WGS), have enabled the precise monitoring of BP. Recently, a multi-level genome typing (MGT) method has been developed for BP. This method involves a sequence of MLST methods in which the number of loci is progressively increased (Payne et al., 2023). MGT involves five levels of resolution for BP and can distinguish closely related strains at the smallest scale. After the introduction of acellular pertussis (aP) vaccination, BP strains lacking vaccine antigens, especially prn, have been widely detected (Barkoff and He, 2019). The genotypes of Chinese epidemic BP strains were different from those of vaccine strains (Fu et al., 2023). In the last decade, several countries have reported adaptive alterations in the vaccine-associated genes of circulating BP strains (Saadatian-Elahi et al., 2016). For example, the most common contemporary ptxA allele is ptxA1, which differs from the ptxA2 allele commonly observed in the vaccine strain. In several countries, the circulating BP strains have evolved to present a non-vaccine antigen genotype (ptxA1/prn2/ptxP3). Meanwhile, vaccines are not available for the pathogenicity of prn-deficient and fim-deficient isolates. These isolates exhibit good fitness to vaccine-induced selection pressure (Barkoff et al., 2018; Carriquiriborde et al., 2019). The Chinese pertussis isolates were reported to primarily harbor the ptxP1 allele (Wang and He, 2015). However, one study demonstrated that mutant strains with the ptxA1/prn2/ptxP3 combination were prevalent in China, accounting for 44.8 and 47.7% of isolates in Shenzhen (Zhang et al., 2020) and Shanghai, respectively (Fu et al., 2024).
Erythromycin (ERY), a macrolide antibiotic, is the preferred antibiotic for preventing and treating pertussis. Two mechanisms of ERY resistance have been identified. The first mechanism involves the acquisition of the ERY-resistant methyltransferase gene (erm), which confers high levels of resistance (Pechère, 2001). Meanwhile, the second mechanism involves mutations in the 23S rRNA gene, which result in structural alterations, impairing the binding of ERY (Bartkus et al., 2003). Macrolide-resistant BP strains, which were first identified in Shandong Province in 2011, have been widely reported in China (Zhang et al., 2013) with incidence rates of 48.6% (51/105), 72.4% (205/283), and 78.13% (25/32) in Shenzhen (Zhang et al., 2020), Shanghai (Fu et al., 2024), and Beijing (Li et al., 2019), respectively.
Until now, a series of studies about BP strains were reported in China, However, there is inadequate study on the BP pandemic strain in Hebei Province. In order better comprehend the evolutionary traits, and molecular characterization of BP in Hebei, as well as the antimicrobial susceptibility. In this study, 62 BP isolates were collected from Hebei between 2019 and 2020. We systematically analyzed the molecular characteristics, the genomic evolution, and the antimicrobial resistance profiles of those strains.
2 Materials and methods 2.1 Bacterial isolatesThis epidemiological cross-sectional study was performed from 2019 to 2020, and it was approved by the Ethics Committee of the Hebei Province Center for Disease Prevention and Control Review Board (no. 2020–35). Three hundred ninety-seven pertussis positive patients were diagnosed by polymerase chain reaction (PCR) analysis of nasopharyngeal swabs from suspected patients at the surveillance site in Hebei Province. The collection was performed according to the Chinese clinical case diagnostic criteria for pertussis. The samples were then spread onto charcoal agar plates supplemented with 10% defibrinated sheep blood and cephalexin and incubated in a humidified incubator at 35°C for 3–7 days. The presence of BP was verified using a matrix-assisted laser desorption/ionization time-of-flight mass spectrometer (Zybio, China). In total, 62 clinical isolates identified as BP were subcultured on a carbon blood agar plate (OXOID, UK) without cefalexin and stored in a −80°C refrigerator (Thermo, USA) for subsequent use.
2.2 Antimicrobial susceptibility testingMinimum inhibitory concentrations (MICs) of four β-lactam antibiotics [amoxicillin (AMX), meropenem (MEM), ceftriaxone (CRO), and cefotaxime (CTX)], two macrolide antibiotics [ERY and azithromycin (AZM)), one lincosamide antibiotic (clindamycin (CLI)], one quinolone antibiotic [levofloxacin (LEV)], and one sulfonamide antibiotic [sulfamethoxazole (TMZ-SMP)] were determined using the E-test strip (BIO-KONT, China). Culture-positive BP isolates were diluted to match a 0.5 McFarland standard and transferred onto a charcoal agar with 10% sheep blood without cephalexin. Antibiotic-free pills were added to the agar to assess growth and purity. Streptococcus pneumoniae (ATCC46916) served as controls. The MICs of antibiotics in the E-test strips were determined using the agar 5 days after inoculation. The Clinical and Laboratory Standards Institute (CLSI) and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) have not established the specific sensitivity and resistance thresholds for BP. Based on pertinent research, an MIC of ≥32 mg/mL was considered as an indication of macrolide resistance. Other antibiotics were evaluated for determining susceptibility by using the breakpoints specified by the 34th edition of the CLSI for Haemophilus influenzae (Yang et al., 2015).
2.3 DNA extraction and WGSThe genomic DNA of the culture-positive isolates was extracted using the QIAamp DNA mini kit (QIAGEN, Germany), following the manufacturer’s instructions. The purity and concentration of the DNA were assessed using an ultralow volume spectrometer (MIULab, China) and an Invitrogen Qubit 4.0 fluorometer (Thermo, USA), respectively. The DNA samples with qualified concentration and purity were randomly sheared into fragments of approximately 350 bp using ultrasonication. The samples were sequenced using Illumina NovaSeq PE 150.
2.4 Single nucleotide polymorphism (SNP) and phylogenetic analysesThe Chinese vaccine strain CS (accession no. GCA_023612135.1) and the international reference strain Tohama I (accession no. GCA_000195715.1) were used as the genome reference sequence. kSNP version 4.1 (Hall and Nisbet, 2023) was used to obtain the alignment of 1,476 SNPs. The number of shared SNPs among 62 BP isolates was 1,269 and was determined based on kmer with an optimum value of 19. The IQ-TREE (Minh et al., 2020) software was used to construct phylogenetic trees based on the maximum-likelihood method with 1,000 ultrafast bootstrap replicates and the model “TN + F + ASC + R3,” which was automatically selected as the best fit. The phylogenetic tree was visualized using TVBOT (Xie et al., 2023).
2.5 MLST, MAST, 23S rRNA genes, and Erm genesMLST of BP was performed by analyzing the following seven specific housekeeping genes using mlst version 2.11 (GiHub, 2022b) to obtain corresponding allele numbers and types: adk, fumC, glyA, tyrB, icd, pepA, and pgm. Previously reported different genotype sequences of antigens were retrieved from GenBank and analyzed using HS-BLASTN 2.6.0 (Chen et al., 2015) to type the vaccine antigen genes ptxP, ptxA, prn, fim2, and fim3 and screen 23S rRNA mutations and erm presence. The sequences were compared with those curated at the BIGSdb-Pasteur platform (Jolley et al., 2018). CS and Tohama I were included as positive controls.
2.6 MGTThe MGT_reads2alleles pipeline (GiHub, 2022a) was applied to extract the alleles from Illumina paired end reads. Genomes that passed the filters were used for further analysis (genome length: 3.5–5 Mb). The generated allele files were uploaded to MGTdb to obtain the MGT assignment. The genome of Tohama I served as the reference genome.
2.7 Public genome datasetThis study compared 375 published genomes of Chinese BP strains and global BP isolates. The genomes in the public dataset were sequenced for various purposes and were from 28 countries and eight geographical zones (Appendix Table 1). Unprocessed short-read sequencing data were obtained from the National Center for Biotechnology Information (NCBI) Sequence Read Archive. The reads were processed using Trimmomatic 0.39 (Bolger et al., 2014) and subjected to de novo assembly using Shovill 1.1.0 (GiHub, 2020).
3 Results 3.1 Laboratory test results and epidemiological characteristics analysisSixty-two strains of BP were isolated from nasopharyngeal swabs of 397 children, resulting in a positive percentage of 15.62% (62/397). The vaccination history of the 62 cases was not available. The age range of children with pertussis spans from 1 month to 9 years, exhibiting a male to female ratio of 1:1. Among these, 9 cases (14.52%) are under 3 months, 18 cases (29.03%) are between 3 and 6 months, 9 cases (14.52%) are between 6 and 18 months, 7 cases (11.29%) are between 18 and 36 months, and 19 cases (30.64%) are over 36 months.
3.2 Antimicrobial susceptibility and 23S rRNA A2047G mutation analysesThe E-test revealed 56 ERY and CLI-resistant (MIC >32 mg/mL) isolates, 55 AZM-resistant (MIC >256 mg/mL) isolates, one ERY and CLI-resistant (MIC >32 mg/mL) but AZM-sensitive (MIC = 16 mg/mL) strain (accession no. GCA_039698275.1), 58 TMZ-SMP-sensitive (MIC range = 0.008–1 mg/mL) isolates, and 53 CTX-sensitive (MIC range = 0.064–2 mg/mL) isolates. All isolates were sensitive to AMX, MEM, and CRO (MIC range = 0.064–2 mg/mL). The erm gene was not detected in any of the genomes. The A2047G mutation in the 23S rRNA gene was detected in 56 strains tested for sensitivity to ERY (MIC >32 mg/mL) using the E-test. The mutation was not detected in the remaining isolates. Figure 1 shows antimicrobial susceptibility and 23S rRNA mutation analysis results.
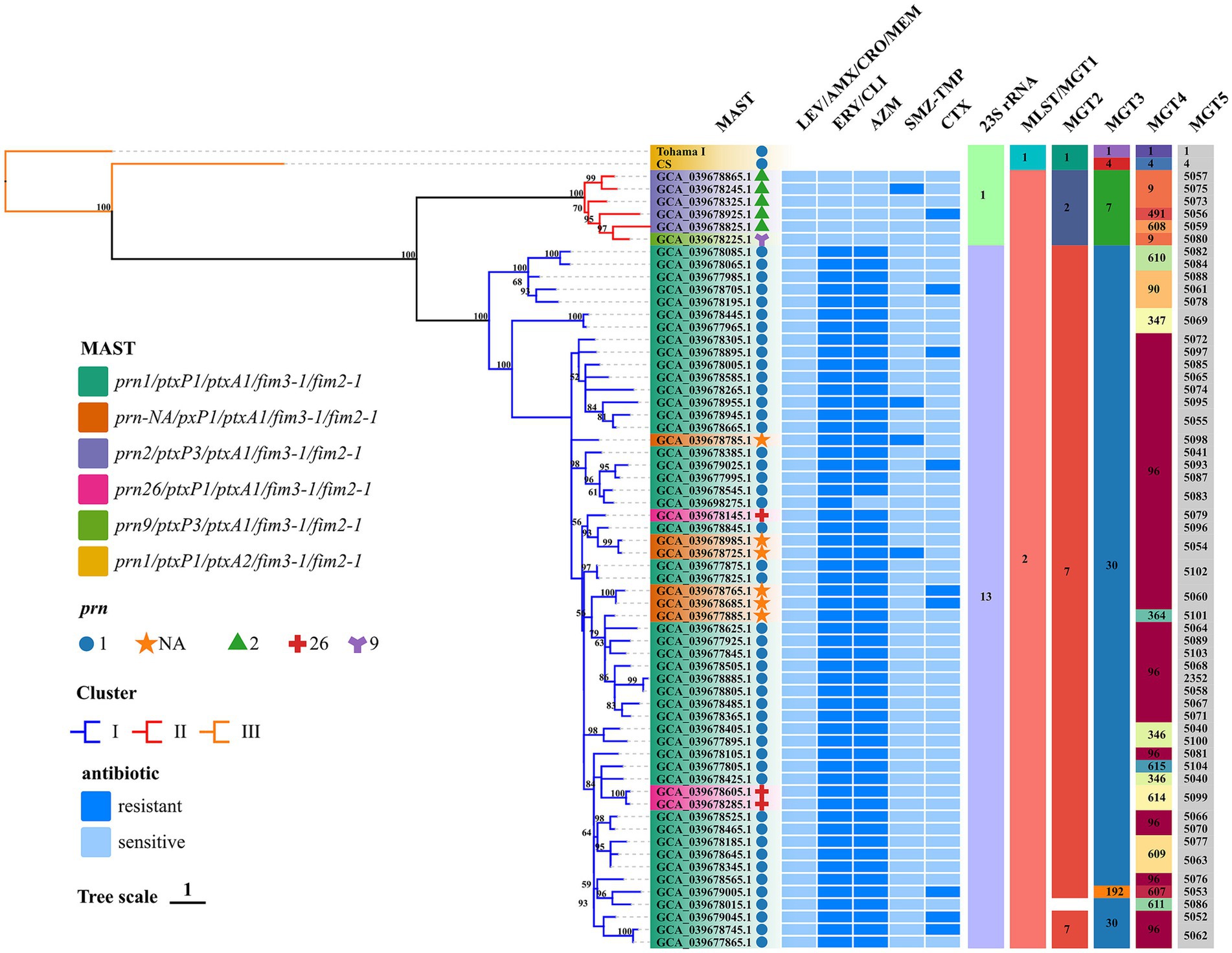
Figure 1. Phylogenomic relationship of 62 Chinese BP isolates. Maximum clade credibility phylogenetic tree for Hebei Province BP isolates in the present studies based on whole-genome SNPs, which included 1,269 SNPs to illustrate the genetic relationship of Hebei Province clinical isolates. Tohama I was used as the outgroup. The ERY resistant lineages I and ERY sensitive lineages II were marked by blue and red, respectively. Bootstrap values of ≥50% were marked at each branch. Prn was represented by graphics of different colors and shapes. Various type details of isolates (MLST, MAST, MGT, phenotype of antimicrobial susceptibility and genotype of BP) were shown as color codes per the legends which are on the left side of the figure, Bootstrap values greater than or equal to 50 were shown in numerical form in the figure MGT3 blank space indicates unassigned, the proportion of branch length is expressed as 1.
3.3 MLST, MAST, MGT, and phylogenetic analysisIn silico analysis the results of BP were consistent with the BIGSdb-Pasteur results. The distribution of types and analysis results are shown in Figure 1. All test isolates had the ptxA1/fim2-1/fim3-1 allele. The ptxP1 and ptxP3 alleles were detected in 90.32% (n = 56) and 9.68% (n = 6) of the isolates, respectively. The following four prn allele types were detected: prn1, prn2, prn9, and prn26. The predominant prn allele was prn1, accounting for 79.03% of the prn alleles (n = 49). Additionally, six isolates exhibited a prn-negative phenotype. The following five antigenic genotypes were detected in the isolates according to prn and ptxP alleles: prn1/ptxP1/ptxA1/fim3-1/fim2-1 (75.81%; n = 47), prn-NA/pxP1/ptxA1/fim3-1/fim2-1 (9.68%; n = 6), prn2/ptxP3/ptxA1/fim3-1/fim2-1 (8.06%; n = 5), prn26/ptxP1/ptxA1/fim3-1/fim2-1 (4.84%; n = 3), and prn9/ptxP3/ptxA1/fim3-1/fim2-1 (1.61%, n = 1). The genotype of the reference strains was prn1/ptxP1/ptxA2/fim3-1/fim2-1. All test isolates belonged to MLST sequence type 2 (ST2) and MGT1 ST2. Additionally, two MGT2 STs (MGT2 ST7 and ST2) and three MGT3 STs (MGT3 ST30, ST7, and ST192) were identified. In this study, 13 and 52 types were identified at the MGT4 and MGT5 levels, respectively. One BP (accession no. GCA_039678015.1) was identified as the unassigned type at the MGT3 level. The phylogenetic tree generated using SNPs revealed the following two lineages: Lineage I (56 isolates) and Lineage II (6 isolates). Among the 62 strains, 1,269 shared SNPs were identified. Of these 1,269 SNPs, 373 (29.39%) were core SNPs. The whole-genome sequences of 62 strains were uploaded to the NCBI Sequence Read Archive (BioProject: PRJNA1110622).
3.4 Correlation between antimicrobial susceptibility, genotypes, and phylogenetic treeThe antigenic genotypes of all ERY-resistant BP and CLI-resistant BP strains were prn1 or prn26 or prn-NA/ptxP1/ptxA1/fim3-1/fim2-1 (88.71, n = 55). In addition, the antigenic genotype of the sensitive strains was prn9 or prn2/ptxP3/ptxA1/fim3-1/fim2-1 (11.29%; n = 7). The antigenic genotypes of TMZ-SMP-resistant BP were as follows: prn-NA/pxP1/ptxA1/fim3-1/fim2-1 (50%; n = 2); prn1/ptxP1/ptxA1/fim3-1/fim2-1 (25%; n = 1), and prn2/ptxP3/ptxA1/fim3-1/fim2-1 (25%; n = 1). ERY-resistant BP belonged to Lineage I, while ERY-sensitive BP belonged to Lineage II. Comparative analysis of several types revealed that 62 BP isolates were consistent and belonged to MLST ST2 and MGT1 ST2. ptxP1 was mostly associated with MGT2 ST7 in the 56 strains with 23S rRNA allele 13 harboring the A2047G mutation. Furthermore, this mutation did not occur in six strains with 23S rRNA that had allele 1, and ptxP3 was mostly linked to MGT2 ST2. ptxP1-ERY-resistant BP strains belonged to MGT3 ST30 (98.21%; n = 55), while ptxP3-ERY-sensitive BP strains belonged to Lineage II and MGT3 ST7. At the MGT4 level, all ptxP3-ERY-sensitive BP strains belonged to MGT4 ST9 (66.67%; n = 4), MGT4 ST491 (16.67%; n = 1), and MGT4 ST608 (16.67%; n = 1). These strains can be separated from ptxP1-ERY-resistant BP strains. Although prn-negative strains could be detected at the MGT4 and MGT5 levels, they could not be distinguished at the MGT1, MGT2, and MGT3 levels and hence belonged to MGT1 ST2, MGT2 ST7, and MGT3 ST30. The STs at the MGT4 level were MGT4 ST96 (83.33%; n = 5) and MGT4 ST364 (16.67%; n = 1). Meanwhile, the STs at the MGT5 level were MGT5 ST5054 (33.33%; n = 2), MGT5 ST5060 (33.33%; n = 2), MGT5 ST5098 (16.67%; n = 1), and MGT5 ST5101 (16.67%; n = 1). At the MGT5 level, the STs of four strains of TMZ-SMP-resistant BP were ST5095, ST5054, ST5075, and ST5098. The STs of nine CTX-resistant strains were ST5052, ST5053, ST5056, ST5061, ST5062, ST5093, and ST5097.
3.5 Phylogenetic tree for the correlation between Hebei and global BP isolatesThe genomes of all 375 strains (62 BP strains from this study and 313 strains from the public dataset of global isolates) were matched using the genome of Tohama I as the outgroup with KSNP4. In total, 10,494 SNPs, including 3,897 core SNPs, were identified. Additionally, 56 strains of ptxP1-ERY-resistant BP from Hebei were closely related to other isolates from Eastern Asia, including China, several cities, and one Japan BP (GenBank accession no. GCA_019974435.1). However, these 56 strains differed from other international strains isolated from the United States, Europe, Australia, Africa, Iran, Israel, Argentina, Brazil, and other regions and countries (Figure 2). Six strains of ptxP3-ERY-sensitive BP from Hebei were closely linked to other isolates from Europe, Latin America, and China (Beijing and Chongqing). Among 375 BP strains, the strains with the 23S rRNA allele A2047G mutation were evolutionary branches from Eastern Asia that were closely connected to 56 strains of ptxP1-ERY-resistant BP. Compared with the BP reference strain Tohama I harboring ptxP1 and ptxA2, the Hebei isolates exhibited a diverse evolution of ptxP and ptxA alleles, while the prn genes underwent varying degrees of deletion.
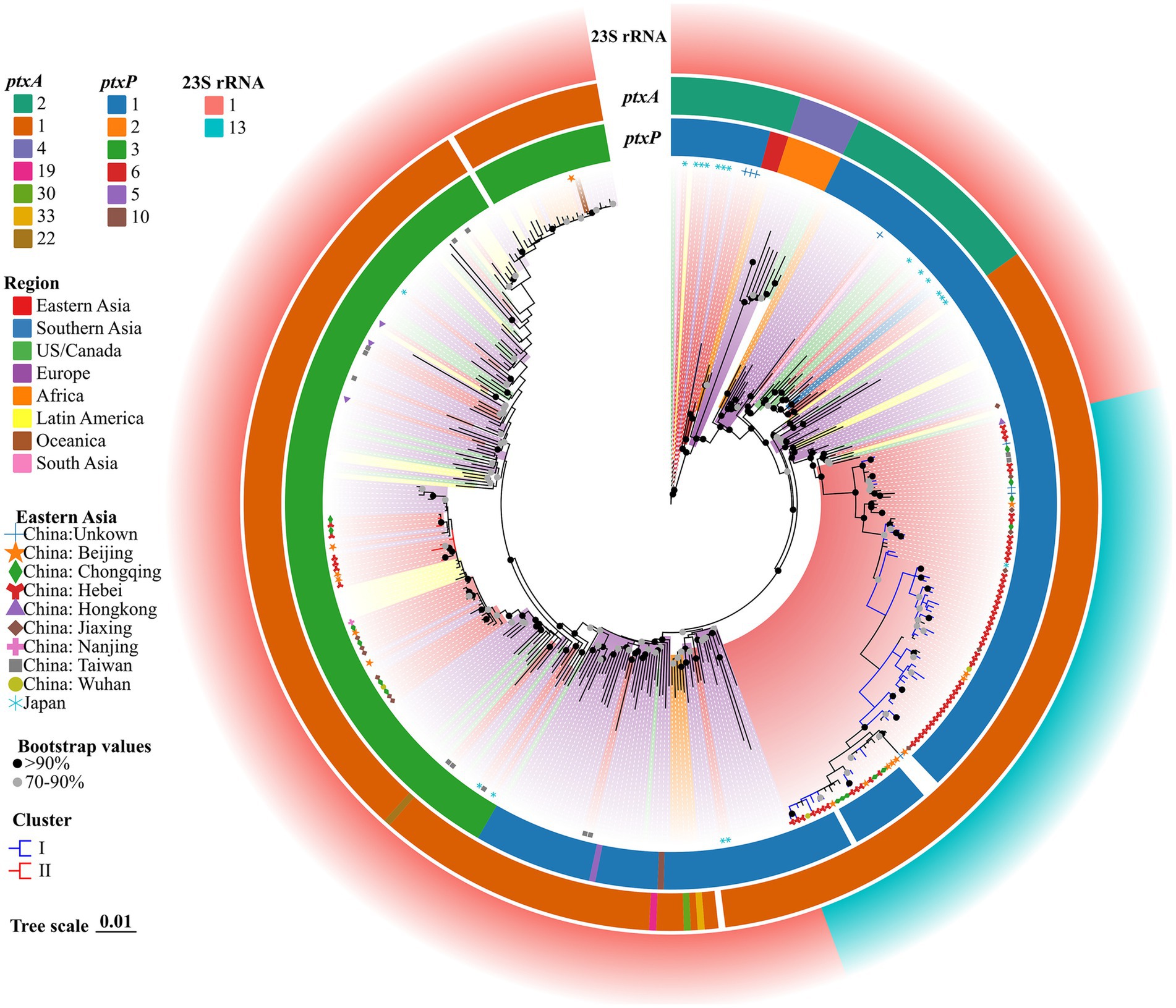
Figure 2. ML phylogenetic tree of 62 Hebei, China, and 313 global BP strains, 2019–2020. Tohama I was used as the outgroup. The ERY resistant lineages I and ERY sensitive lineages II in this study were marked by blue and red, respectively. The BP of eight regions were represented in different colors. Regions in Eastern Asia were represented by graphics of different shapes and colors. Isolate details (ptxA, ptxP and 23S rRNA allele type) were shown as color codes as per the legends. The blank space in the figure represents the gene deletion corresponding to the BP. Bootstrap values greater than or equal to 70 were shown in the figure as circles of two different colors, the proportion of branch length was expressed as 0.01.
4 DiscussionActive monitoring of pertussis has revealed that the incidence rate of pertussis is increasing in Hebei Province (Zheng et al., 2024). Therefore, there is a need to determine the cause of increased prevalence rates of pertussis. Previous studies have demonstrated that the resurgence of pertussis can be attributed to BP isolates harboring antigen gene mutations and their distinct characteristic relative to the vaccine strain (Miyaji et al., 2013). To understand the potential causes of pertussis recurrence in Hebei, this study investigated the genetic diversity and molecular epidemiological characteristics of 62 BP strains collected between 2019 and 2020.
ERY-based macrolide antibiotics are the first treatment choice for pertussis (Zhang et al., 2022). In this study, antibiotic sensitivity analysis of 62 strains of BP isolates from Hebei Province revealed that Hebei isolates exhibited high resistance to macrolides, Allele 13 of the 23S rRNA locus which harbored the A2047G mutation was unique (Bridel et al., 2022) for ERY-resistant BP. Additionally, ptxP1-ERY-sensitive BP strains harbored the 23S rRNA A2047G mutation, and ptxP3-ERY-sensitive BP strains did not harbor the A2047G mutation. The incidence rates of ERY and AZM resistance were more than 90%. This indicates that ptxP1-ERY-sensitive BP strains harboring A2047G mutation were widely distributed in Hebei. Hence, the high incidence rate of macrolide resistance among BP strains in China must be addressed. The incidence rate of CLI resistance was 90.32%. CLI belongs to the lincosamide class of antibiotics with similar mechanisms as macrolides. Thus, the incidence of CLI resistance was like that of ERY resistance. One isolation in this study (GCA_039698275.1) was resistant to ERY and CLI (MIC >32 mg/mL) but sensitive to AZM (MIC = 16 mg/mL). Previous studies have reported two strains that were resistant to ERY and AZM but sensitive to clarithromycin (Zhang et al., 2022). The 23S rRNA A2047G mutation was assumed to be the primary etiological factor for the resistance of BP to macrolides (Barkoff and He, 2019). However, the precise mechanism underlying the variations in ERY and AZM resistance has not been elucidated. SMZ-TMP was traditionally considered the secondary therapeutic option (Zhang et al., 2020). In this study, the incidence rate of SMZ-TMP sensitivity was 93.55%. In patients with pertussis exhibiting resistance to macrolides, SMZ-TMP must be recommended for the treatment of pertussis caused by macrolide-resistant strains in children aged over 2 months in China (Group et al., 2024). AMX, MEM, CRO, and CTX belong to β-lactam antibiotics. All 62 BP isolates were sensitive to AMX, MEM, and CRO. However, the incidence rate of CTX sensitivity was 85.48%. This suggested that these four drugs can be used to treat patients with pertussis. Early studies did not support the use of β-lactam antibiotics to treat pertussis. However, some studies suggested the effectiveness of β-lactam antibiotics in pertussis. The in vitro sensitivity to different β-lactam drugs may significantly vary (Yao et al., 2024). In this study, 62 BP strains were sensitive to the quinolone drug LEV. However, LEV exerts toxic effects in children and can be used to treat only teenagers or adults (Yao et al., 2024). Further studies are needed to assess the viability of β-lactam antibiotic treatment for the clinical management of patients, especially infants and young children, infected with macrolide-resistant strains. Furthermore, the effectiveness of quinolone antibiotics must be evaluated through clinical trials.
Globally, ptxP3 strains are commonly detected. In contrast, this study revealed that ptxP1-Macrolide-resistant BP strains were common in Hebei with 90.32% of the isolates harboring the ptxP1 allele (Barkoff and He, 2019; Xu et al., 2019). The prevalence of ptxP1-Macrolide-resistant BP strains in China may be attributed to antibiotic selection. The ptxP1-Macrolide-resistant BP strains have several advantages over ptxP3-Macrolide-sensitive BP strains in the population in which the usage of antibiotics is high. The MAST analysis categorized 62 strains of BP into six distinct categories with most strains (75.81%) exhibiting the antigen genotype prn1/ptxP1/ptxA1/fim3-1/fim2-1. Both the Chinese vaccination strain CS and the international strain Tohama I harbored the prn1/ptxP1/ptxA2/fim3-1/fim2-1 genetic markers. All BP samples in this study contained the ptxA1/fim3-1/fim2-1 genotype. However, in this study, all BP isolates carried the ptxA1/fim3-1/fim2-1 alleles, while both prn and ptxP exhibited distinct variations in their alleles. Furthermore, the prn gene was demonstrated to exhibit a range of allelic variations in this study. The polymorphism of prn gene and the transmission of prn-defective isolates may represent the selective avoidance of vaccine-induced immunity and antibiotics by bacteria (Xu et al., 2019). prn deficiency has been documented in several areas when ACV containing prn was used. In particular, the incidence of prn-deficient strains was high in Australia and some other nations (Barkoff and He, 2019). Six prn-deficient strains were identified in this research and there had studies discovered that prn-negative strains exhibit increased adaptation in mouse models (Safarchi et al., 2015). Studies have shown that prn-deficient strain was the most suitable genotype in the era of aP vaccine and after the introduction of aP vaccine, the average fitness of prn-deficient strain was 1.26 times than those strains which carrying prn gene (Lefrancq et al., 2022), so it is expected that the frequency of prn-deficient strains will further increase in the future.
The MLST and MAST of Hebei isolates differed from those of vaccination strains. However, the resolution was low. To differentiate between closely related isolates in the BP population, the genome must be widely covered due to its limited genetic diversity. SNP-based evolutionary trees can provide an accurate representation of phylogenetic evolution and linkages. This study revealed three lineages, which included the reference strain. Furthermore, the strains found in Hebei Province shared a distant genetic link with the reference strain, indicating that the epidemic strain has undergone varied degrees of mutation when compared with the vaccination strain. Compared with the international epidemic BP strain, 56 strains of ptxP1-ERY-resistant BP formed a distinct branch in the Eastern Asia region where China is located. Meanwhile, six strains of ptxP3-ERY-sensitive BP were closely related to individual strains in parts of Europe and economically developed cities in China. MGT supports multi-level resolution. The MGT5 level (BP core gene MLST) has the same resolution as an existing entire gene. MLST scheme maintains the standardization that is inherent in a cgMLST scheme (Payne et al., 2023). MGT1 and MGT2 were compatible with the MLST and BPagST results, respectively. The resolution of BP genotyping was significantly high at the MGT4 and MGT5 levels, which can be used to identify outbreaks and track their origins.
In conclusion, the epidemic trend of antimicrobial susceptibility of BP in Hebei Province is consistent with that of BP in China in recent years (Li et al., 2019; Xu et al., 2019; Zhang et al., 2022; Fu et al., 2024). The antigen genotype of circulating BP stains in Hebei Province is consistent with the epidemic trend of BP in Midwest China in recent years (Xu et al., 2019; Zhang et al., 2022) but varied from the epidemic trend of BP in the southern part of the relatively developed regions, such as Shenzhen (Zhang et al., 2020) and Shanghai (Fu et al., 2024), and industrialized nations, including Australia and the United States (Barkoff and He, 2019). Generally, ptxP3-BP is more common in countries that have used ACV than ptxP1-BP, which is common in countries that have used WCV. WCV was replaced in China in 2012 and has been the sole vaccine used in China since 2013. The implementation of WCV in China was delayed by a decade when compared with that in industrialized countries, such as the United States and Australia. WCV is implemented in developed areas of China, such as Shanghai but is less commonly implemented in underdeveloped areas. Thus, ptxP3 strains may not have had enough time to replace ptxP1 strains in China (Xu et al., 2019). This can explain the differential circulating strains between some Chinese cities. Compared with those of BP vaccination strains, important virulence protein genes and core genomes of Hebei isolates have significantly evolved. The findings of this study reveal potential gaps in BP genetic diversity in Hebei Province and provide essential reference values for improving the pertussis vaccine in China. Nonetheless, the current findings may be constrained by the limited number of strains examined, and subsequent research should encompass a broader range and greater volume of sampling to validate the conclusions. Furthermore, thorough screening for potential mechanisms of antibiotic resistance commonly associated with individuals suffering from pertussis is necessary, and thereby elucidate the relationship between the development of antimicrobial resistance in BP and alterations in genotypes.
Data availability statementThe datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statementThe studies involving humans were approved by Hebei Province Center for Disease Prevention and Control Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributionsBH: Project administration, Writing – review & editing. ZJ: Supervision, Writing – review & editing. FZ: Writing – original draft, Writing – review & editing. WZ: Data curation, Writing – review & editing. SD: Data curation, Writing – review & editing. LW: Formal analysis, Writing – review & editing. HaZ: Software, Writing – review & editing. HoZ: Methodology, Validation, Writing – review & editing. RW: Writing – review & editing, Data curation. YG: Formal analysis, Supervision, Writing – review & editing. YS: Project administration, Resources, Supervision, Writing – review & editing.
FundingThe author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants from the Hebei Provincial Health Commission Project (20210177), People’s Republic of China.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary materialThe Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1498638/full#supplementary-material
Footnotes ReferencesBarkoff, A. M., and He, Q. (2019). Molecular epidemiology of Bordetella pertussis. Adv. Exp. Med. Biol. 1183, 19–33. doi: 10.1007/5584_2019_402
Crossref Full Text | Google Scholar
Barkoff, A. M., Mertsola, J., Pierard, D., Dalby, T., Hoegh, S. V., Guillot, S., et al. (2018). Surveillance of circulating Bordetella pertussis strains in Europe during 1998 to 2015. J. Clin. Microbiol. 56, 10–1128. doi: 10.1128/JCM.01998-17
PubMed Abstract | Crossref Full Text | Google Scholar
Bartkus, J. M., Juni, B. A., Ehresmann, K., Miller, C. A., Sanden, G. N., Cassiday, P. K., et al. (2003). Identification of a mutation associated with erythromycin resistance in Bordetella pertussis: implications for surveillance of antimicrobial resistance. J. Clin. Microbiol. 41, 1167–1172. doi: 10.1128/JCM.41.3.1167-1172.2003
PubMed Abstract | Crossref Full Text | Google Scholar
Bridel, S., Bouchez, V., Brancotte, B., Hauck, S., Armatys, N., Landier, A., et al. (2022). A comprehensive resource for Bordetella genomic epidemiology and biodiversity studies. Nat. Commun. 13:3807. doi: 10.1038/s41467-022-31517-8
PubMed Abstract | Crossref Full Text | Google Scholar
Carriquiriborde, F., Regidor, V., Aispuro, P. M., Magali, G., Bartel, E., Bottero, D., et al. (2019). Rare detection of Bordetella pertussis pertactin-deficient strains in Argentina. Emerg. Infect. Dis. 25, 2048–2054. doi: 10.3201/eid2511.190329
PubMed Abstract | Crossref Full Text | Google Scholar
Fu, P., Zhou, J., Meng, J., Liu, Z., Nijiati, Y., He, L., et al. (2023). Emergence and spread of MT28 ptxP3 allele macrolide-resistant Bordetella pertussis from 2021 to 2022 in China. Int. J. Infect. Dis. 128, 205–211. doi: 10.1016/j.ijid.2023.01.005
PubMed Abstract | Crossref Full Text | Google Scholar
Fu, P., Zhou, J., Yang, C., Nijiati, Y., Zhou, L., Yan, G., et al. (2024). Molecular evolution and increasing macrolide resistance of Bordetella pertussis, Shanghai, China, 2016–2022. Emerg. Infect. Dis. 30, 29–38. doi: 10.3201/EID3001.221588
PubMed Abstract | Crossref Full Text | Google Scholar
Group, P. I., Children, N., and China Clinical Practice Guidelines Alliance Methodology, C. (2024). Guidelines for diagnosis and management and prevention of pertussis of China (2024 edition). Chin. Med. J. 104, 1258–1279. doi: 10.3760/cma.j.cn112137-20240124-00179
PubMed Abstract | Crossref Full Text | Google Scholar
Hall, B. G., and Nisbet, J. (2023). Building phylogenetic trees from genome sequences with kSNP4. Mol. Biol. Evol. 40:msad235. doi: 10.1093/molbev/msad235
Crossref Full Text | Google Scholar
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST. Org website and their applications. Wellcome Open. Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
PubMed Abstract | Crossref Full Text | Google Scholar
Lefrancq, N., Bouchez, V., Fernandes, N., Barkoff, A.-M., Bosch, T., Dalby, T., et al. (2022). Global spatial dynamics and vaccine-induced fitness changes of Bordetella pertussis. Sci. Transl. Med. 14:eabn3253. doi: 10.1126/scitranslmed.abn3253
PubMed Abstract | Crossref Full Text | Google Scholar
Li, L., Liu, Y., Jia, J., Yuan, L., Shi, W., Meng, Q., et al. (2019). Antimicrobial susceptibility and antigen genotypes of Bordetella pertussis strains isolated from neonates. Chin. J. Contemp. Pediatr. 21, 208–213. doi: 10.7499/j.issn.1008-8830.2019.03.004
PubMed Abstract | Crossref Full Text | Google Scholar
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
PubMed Abstract | Crossref Full Text | Google Scholar
Miyaji, Y., Otsuka, N., Toyoizumi-Ajisaka, H., Shibayama, K., and Kamachi, K. (2013). Genetic analysis of Bordetella pertussis isolates from the 2008–2010 pertussis epidemic in Japan. PLoS One 8:e77165. doi: 10.1371/journal.pone.0077165
PubMed Abstract | Crossref Full Text | Google Scholar
Payne, M., Xu, Z., Hu, D., Kaur, S., Octavia, S., Sintchenko, V., et al. (2023). Genomic epidemiology and multilevel genome typing of Bordetella pertussis. Emerg. Microbes Infect. 12:2239945. doi: 10.1080/22221751.2023.2239945
PubMed Abstract | Crossref Full Text | Google Scholar
Pechère, J. C. (2001). Macrolide resistance mechanisms in gram-positive cocci. Int. J. Antimicrob. Agents 18, 25–28. doi: 10.1016/s0924-8579(01)00407-1
Crossref Full Text | Google Scholar
Saadatian-Elahi, M., Plotkin, S., Mills, K. H. G., Halperin, S. A., McIntyre, P. B., Picot, V., et al. (2016). Pertussis: biology, epidemiology and prevention. Vaccine 34, 5819–5826. doi: 10.1016/j.vaccine.2016.10.029
Crossref Full Text | Google Scholar
Safarchi, A., Octavia, S., Luu, L. D. W., Tay, C. Y., Sintchenko, V., Wood, N., et al. (2015). Pertactin negative Bordetella pertussis demonstrates higher fitness under vaccine selection pressure in a mixed infection model. Vaccine 33, 6277–6281. doi: 10.1016/j.vaccine.2015.09.064
PubMed Abstract | Crossref Full Text | Google Scholar
Wang, Z., and He, Q. (2015). Bordetella pertussis isolates circulating in China where whole cell vaccines have been used for 50 years. Clin. Infect. Dis. 61, 1028–1029. doi: 10.1093/cid/civ457
PubMed Abstract | Crossref Full Text | Google Scholar
Xie, J., Chen, Y., Cai, G., Cai, R., Hu, Z., and Wang, H. (2023). Tree visualization by one table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 51, W587–W592. doi: 10.1093/nar/gkad359
PubMed Abstract | Crossref Full Text | Google Scholar
Xu, Y., Tan, Y., Asokanathan, C., Zhang, S., Xing, D., and Wang, J. (2015). Characterization of co-purified acellular pertussis vaccines. Hum. Vaccin. Immunother. 11, 421–427. doi: 10.4161/21645515.2014.988549
PubMed Abstract | Crossref Full Text | Google Scholar
Xu, Z., Wang, Z., Luan, Y., Li, Y., Liu, X., Peng, X., et al. (2019). Genomic epidemiology of erythromycin-resistant Bordetella pertussis in China. Emerg. Microbes Infect. 8, 461–470. doi: 10.1080/22221751.2019.1587315
PubMed Abstract | Crossref Full Text | Google Scholar
Yang, Y., Yao, K., Ma, X., Shi, W., Yuan, L., and Yang, Y. (2015). Variation in Bordetella pertussis susceptibility to erythromycin and virulence-related genotype changes in China (1970-2014). PLoS One 10:e0138941. doi: 10.1371/journal.pone.0138941
PubMed Abstract | Crossref Full Text | Google Scholar
Yao, K., Meng, Q., Shi, W., Yuan, L., and Hu, Y. (2024). Thoughts on the selection of antimicrobials for current pertussis treatment in China. Chin. Clin. J. Pract. Pediatr. 39, 85–88. doi: 10.3760/cma.j.cn101070-20231128-00385
Crossref Full Text | Google Scholar
Zhang, Q., Li, M., Wang, L., Xin, T., and He, Q. (2013). High-resolution melting analysis for the detection of two erythromycin-resistant Bordetella pertussis strains carried by healthy schoolchildren in China. Clin. Microbiol. Infect. 19, E260–E262. doi: 10.1111/1469-0691.12161
PubMed Abstract | Crossref Full Text | Google Scholar
Zhang, J., Wang, H., Yao, K., Liu, Y., Lei, Y., Deng, J., et al. (2020). Clinical characteristics, molecular epidemiology and antimicrobial susceptibility of pertussis among children in southern China. World J. Pediatr. 16, 185–192. doi: 10.1007/s12519-019-00308-5
PubMed Abstract | Crossref Full Text | Google Scholar
Zhang, J., Zhang, D., Wang, X., Wei, X., and Li, H. (2022). Macrolide susceptibility and molecular characteristics of Bordetella pertussis. J. Int. Med. Res. 50:3000605221078782. doi: 10.1177/03000605221078782
PubMed Abstract | Crossref Full Text | Google Scholar
Zheng, F., Sun, Y., Zhang, H., Zhang, H., He, B., Jia, Z., et al. (2024). Epidemiological and spatial-temporal clustering characteristics of pertussis in Hebei Province from 2013 to 2022. Chin. J. Epidemiol. 45, 213–219. doi: 10.3760/cma.j.cn112338-20230811-00064
留言 (0)