Reagents and solvents
All chemicals and solvents were commercially sourced from suppliers Sigma-Aldrich (Bornem, Belgium), Fluka (Bornem, Belgium), Fisher (Doornik, Belgium) and Acros Organics (Geel, Belgium), and were used without further purification. LY2510924 and NOTA-SC were purchased from Pepmic Co., Ltd (Suzhou, China) and were analyzed by LC-HRMS (see Supplementary data). Buffers used for the radiolabeling were treated with Chelex 100 (sodium form, 50–100 mesh, Sigma Aldrich) for 1 h to remove any trace metals. Any buffer and solvent were degassed and filtered prior use.
Radiosynthesis of [18F]AlF-NOTA-SC
The fluorine-18 labeling of NOTA-SC was performed using a custom-made remote-controlled lead-shielded synthesis module. Fluorine-18 was produced on-site using a cyclotron (IBA Cyclone 18/9, IBA, Louvain-la-Neuve, Belgium) by irradiation of H218O with 18-MeV protons [62]. The resulting activity was trapped on an activated Sep-Pak light Accel plus anion exchange cartridge (Cl− form: Waters, Milford, Massachusetts, USA) and rinsed with 4 mL HPCE grade water (Sigma Aldrich, Saint Louis, MO, USA). Subsequently, the 18F-fluoride was eluted with 250 µL of 0.9% NaCl (99.999% trace metals basis NaCl (Sigma Aldrich) in HPCE grade water (Sigma Aldrich)) into a vial containing 100 nmol of AlCl3 in sodium acetate (0.1 M, pH 4.5).
After allowing the mixture to react at room temperature for 5 min to form [18F]AlF, the precursor solution containing 100 nmol of NOTA-SC and 0.95 mg/mL of sodium ascorbate in sodium acetate (0.1 M pH 4.1/ absolute ethanol (30/70 v/v)), was added and allowed to react at 100 °C for 25 min. Following cooling, the crude reaction mixture was diluted with 1 mL of water and injected onto a preparative HPLC column (XBridge BEH OBD Prep, 130 A, 5 μm, 10 × 250 mm) eluted with a mixture of Ammonium acetate 0.05 M pH 5.5 and Ethanol (70/30 V/V) at a flow rate of 2 mL/min. The retention time of [18F]AlF-NOTA-SC was 20 min. The purified radiotracer was diluted with 0.9% w/v NaCl and was sterile filtered using a 0.22 μm filter (Millex-GV, 0,22 μm, PVDF, 13 mm, Merck KGaA, Darmstadt, Germany) resulting in a final batch formulation of < 1% v/v ethanol in 0.9% w/v NaCl solution. The radioactivity of the final product was measured using an ionization chamber (COMECER VIK-203, Comecer S.p.A., Castel Bolognese, Italy).
Analytical HPLC analysis to confirm the identity and to determine the radiochemical and chemical purity and the molar activity was performed on a Hamilton PRP-1 column (1.5 μm, 4.1 × 150 mm, Hamilton, Bonaduz, Graubünden, Switzerland) isocratically eluted with 25% CH3CN in 10 min at 0.75mL/min with the UV detection set at 220 nm.
The radiochemical yield was calculated by dividing the activity measured in the final purified batch by the decay-corrected starting activity of 18F-fluoride obtained from the cyclotron. The apparent molar activity was calculated using a calibration curve with [natF]AlF-NOTA-SC.
[natF]AlF labeling of NOTA-SC
The AlF-labeling of NOTA-SC was conducted following established procedures [39, 62]. Briefly, AlCl3 (490 µL, 5.1 mM in 0.1 M sodium acetate, pH 4.1, 10 eq.) and NaF (10 µL, 10 mg/mL in 0.1 M sodium acetate, pH 4.1, 10 eq.) were added and allowed to react for 5 min at room temperature to form AlF2+. Subsequently, the precursor NOTA-SC (1.0 mg in 500 µL absolute ethanol, 1 eq.) (Mr 1990.29 Da) was added and allowed to react for 30 min at 95 °C. After cooling, the solution was diluted with 20 mL of high-purity water (HPCE, Sigma Aldrich, Saint Louis, MO, USA) and loaded onto a preconditioned Sep-Pak Plus Light C18 cartridge (Waters) to remove non-chelated fluoride species. [natF]AlF-NOTA-SC was then eluted from the cartridge using 0.5 mL acetonitrile and 1 mL water, and filtered through a Captiva PTFE + GF 0.45 μm filter (Agilent Technologies, Santa Clara, CA, USA). The final solution was frozen on dry ice and lyophilized overnight. The expected weight was C95H138AlFN22O25 = 2034.26 Da; the found weight was 2034.24 ± 0.01 Da, which was calculated based on the charged envelope value.
In vitro assessmentCell origin and culture conditions
The human glioblastoma U87.MG and U87.CD4 cells were generously provided by Dr. D. Littman (The Skirball Institute of Biomoleculair Medicine, New York, USA) [63, 64] with U87.MG being the parent cell line for the subsequent transfected U87 cell lines. The GPCR transfection was performed in-house with overexpression of CXCR4 [65], ACKR3 [66] and CCR5 [65] characterized and reported.
U87.MG cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 4 mM L-glutamine, 10% (v/v) fetal calf serum (Thermo Fisher Scientific, Waltham, MA, USA) and 0.01 M HEPES. U87.CD4 cells were supplemented with geneticin (0.2 mg/mL) (Thermo Fisher Scientific), while U87.CD4.CXCR4, U87.CD4.CCR5, and U87.CD4.ACKR3 cell lines additionally received puromycine (2 µg/mL) (Sigma-Aldrich). Jurkat human T-cell leukemia cells (ATCC) endogenously expressing hCXCR4 were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 2 mM glutamine. All the cells were maintained in a humidified incubator at 37 °C with 5% CO2.
The MM.1 S cells (ATCC CRL-2974) were purchased and cultured in RPMI-1640 (Thermo Fisher Scientific) supplemented with 10% (v/v) fetal calf serum (Thermo Fisher Scientific). Each cell line was routinely passaged twice weekly under standard culture conditions of 37 °C and 5% CO2.
Human and murine CXCR4 affinity
The affinity towards mCXCR4 or hCXCR4 has been assessed, alike described by Schoofs et al. [67]. Briefly, 100 µL of the compound of interest dissolved in assay buffer [20 mM HEPES (Thermo Fisher Scientific) and 0.2% BSA (Sigma-Aldrich) dissolved in Hank’s Balanced Salt Solution (Thermo Fisher Scientific) at pH 7.4] was added onto a 96-wells plate in a dilution series ranging from 0.15 nM to 10,000 nM. Then 50 µL containing 0.25∗106 U87.mCXCR4 or Jurkat cells were added to each well followed by a ~ 10 min incubation step. Finally 50 µL (100 ng/mL) of murine or human CXCL12AF647 (Almac, Craigavon, United Kingdom), respectively were added and the cell plate was left to incubate for 30 min at room temperature in the dark. Then the supernatant was changed twice by centrifuging the plate at 400xg for 5 min and finally the cells were fixed in 1% paraformaldehyde (Merck, Darmstadt, Germany). The final quantitative readout of the fluorescence was done via flow cytometry. The IC50 was calculated applying a nonlinear regression in GraphPad Prism 9 (Graph Pad Software, San Diego, CA, USA). Every experiment has been performed at least in quadruplicates.
In vitro cell binding
The binding assays were conducted using 0.2∗106 U87.MG, U87.CD4, U87.CD4.CXCR4, U87.CD4.CCR5, U87.CD4.CXCR7 or MM.1 S cells, which were plated 24 h prior to the experiment. On the day of the assay, the medium was removed and approximately 150 kBq radiotracer (3.8 µL of 376.0 MBq/mL) was added to each well, diluted in assay buffer consisting of 20 mM HEPES (Thermo Fisher Scientific) and 0.2% BSA (Sigma-Aldrich) in Hank’s Balanced Salt Solution (Thermo Fisher Scientific) at pH 7.4. For blocking studies, the corresponding competitor (75 µM) was added simultaneously with the radiotracer: AMD3100 (20.1 µL) (Sigma-Aldrich), Maraviroc (MedChemExpress, Monmouth Junction, NJ, USA), VUF11207 (4.2 µL) (Laboratory of Virology and Chemotherapy, KU Leuven, Belgium), or LY2510924 (2.1 µL) (Pepmic) as self-blocker. Incubation was performed at 37 °C–4 °C for 1 h.
After removing the medium, the cells were washed with 600µL ice cold PBS to collect the “non-bound” fraction. The cells were then washed twice for 5 min each with 600 µL of ice cold glycine-HCl buffer (50 mM, pH 2.8) to collect the “membrane bound” fraction. Next, 500 µL of solvent A100 (Chemometec, Allerod, Denmark) and 500 µL of reagent B (Chemometec) were added to lyse the cells, followed by a wash with 500 µL PBS to collect the “internalized” fraction. Each fraction was then counted in a gamma counter (Perkin-Elmer, Wizard2 2480, Waltham, MA, USA) and the number of cells was determined using a NucleoCounter® NC-100™ (Chemometec). The membrane and internalized fraction were corrected for 0.2∗106 cells. Each experiment was performed in triplicate.
The same methodology was applied to non-adherent MM.1 S cells, with the exception of the washing steps. Here the cells were centrifuged for 5 min at 400xg, and the supernatant was extracted after each step to calculate the different fractions.
In vitro autoradiography
Frozen 20 μm tumor slices were thawed and incubated in 50 mM Tris.HCl (Sigma-Aldrich) at pH 7.4 for 5 min at room temperature. Subsequently, the tumor slices were incubated with 3.43 MBq/mL of the radiotracer diluted in assay buffer containing 20 mM HEPES (Thermo Fisher Scientific) and 0.2% BSA (Sigma-Aldrich) in Hank’s Balanced Salt Solution (Thermo Fisher Scientific) at pH 7.4, for 30 min at room temperature. Receptor specificity was assessed by co-incubating with AMD3100 (75 µM). After removing the radiotracer solution, the tumor slices were washed twice for 5 min with PBS containing 0.3% BSA at 4 °C, followed by a final dip in water. The slices were dried and exposed overnight on a phosphor storage screen (Super resolution screen, Perkin Elmer, Waltham, MA). The read-out was performed using the Cyclone Plus system (Perkin Elmer) and Optiquant software (Perkin Elmer). Percentage block versus control was calculated as (Digital Light Units (DLU)/mm2 in the presence of AMD3100)/(DLU/mm2 tracer only) on three tissue sections.
In vivo assessmentAnimal welfare
The naïve biodistribution study was conducted in four-week-old female NMRI mice (Envigo RMS BV, Venray, The Netherlands), while the tumor model used four-week-old female SCID mice (CB17.Cg-PrkdcscidLystbg-J/Crl, Charles River Laboratories, Sulzfeld, Germany). The non-human primate rhesus macaque (Macaca mulatta) was a 9 years old male.
Mice were individually housed in ventilated cages with monitored temperature and humidity. They had ad libitum access to water and food, under a 12-hour light-dark cycle. The non-human primate was socially housed (two to three animals per cage) in ventilated cages with temperature and humidity monitoring. Food access was ad libitum and water access was given once per day (40 mL/kg). The animals have a normal day-night cycle due to access to natural light.
All animal experiments were approved by the KU Leuven ethical review board (Reference P010/2023 (mice studies) and P112/2019 (non-human primate study)), and were conducted in compliance with Directive 2010/63/EU [68].
Tumor xenograft modelling in mice with U87.CD4, U87.CD4.CXCR4 or MM.1 S cells
The same tumor model was utilized as described by Burke et al. [69]. Briefly, 1.5 × 106 U87.CD4, U87.CD4.CXCR4 or MM.1 S cells were mixed in a 1:1 ratio using Cultrex Basement Membrane Extract (R&D systems, Minneapolis, MN, USA) and inoculated subcutaneously on the shoulder of four-week old female SCID mice (CB17.Cg-PrkdcscidLystbg-J/Crl; Charles River Laboratories). Tumor size was measured every two days using a Vernier caliper and calculated using the formula: height × length × width. Once tumors exceeded 200 mm3 size, mice were randomly assigned to different cohorts for subsequent biodistribution studies.
The systemic MM.1 S model was conducted as described by Hofgaard et al. [70]. Briefly, 5*106 MM.1 S cells dissolved in PBS were injected via the tail vein into four-weeks-old female SCID mice (CB17.Cg-PrkdcscidLystbg-J/Crl; Charles River Laboratories). Tumor progression was monitored by biweekly weight measurements and behavioral assessments. Six weeks post tumor inoculation, the tumor model was ready for radiotracer injections.
Ex vivo autoradiography
Shortly after the ex vivo biodistribution, the tumors were snap-frozen in isopentane (-50 °C). Subsequently, 20 μm tissue slices were cut using a cryotome (Shandon cryotome FSE; Thermo Fisher, Waltham, MA) and mounted on adhesive microscope slides (Superfrost Plus; Thermo Fisher Scientific). These slices were then exposed overnight on a phosphor storage screen (Super-resolution screen; Perkin Elmer).
PET/CT imaging and biodistribution in wild-type and xenografted mice
Female NMRI mice, U87.CD4/U87.CD4.CXCR4 or MM.1 S tumor-bearing mice were anesthetized (2.5% isoflurane in O2 at 1 L/min flow rate) and injected i.v. with [18F]AlF-NOTA-SC (1.5–3.0 MBq/ 0.18 nmol/mouse, n = 4 (NMRI), n = 3 (U87.CD4/U87.CD4.CXCR4 or MM.1 S), either with or without AMD3100 administered i.p. (5 mg/kg 15 min prior to the tracer, n = 1 (MM.1 S)). Dynamic PET images were acquired for 60 min immediately after injection using a β-cube PET scanner (Molecubes, Ghent, Belgium). Throughout the procedure, mice remained under continuous gas anesthesia (2.5% isoflurane in O2 at 1 L/min flow rate), with temperature and respiration monitored.
Following PET scanning, CT imaging was performed for anatomical co-registration using an X-cube CT scanner (Molecubes) using the ‘General Purpose’ protocol with the following parameters: 50 kVp, 480 exposures, 85 ms/projection, 100 µA tube current, rotation time 60 s. After scanning, mice were sacrificed at 75 min p.i., and a whole-body biodistribution study was performed. Blood and major organs were collected in tared tubes and weighed. Quantification of radioactivity in blood, organs, and other body parts was performed using an automated gamma counter equipped with a 3-inch NaI(Tl) well crystal coupled to a multichannel analyzer, mounted in a sample changer (Perkin Elmer 1480 Wizard 3q). Counts were corrected for background radiation, physical decay and counter dead time. The values have been expressed in standardized uptake values ((SUV; tissue activity concentration (MBq/g)/[injected activity (MBq)/body weight (g)])) or percentage of injected dose ((%ID; tissue activity (MBq)/injected activity (MBq))).
[18F]FDG PET/CT imaging in MM.1 S xenografted mice
The [18F]FDG scan was performed as described by Ribeiro et al. [71]. Briefly, MM.1 S tumor-bearing mice were fasted for 12 h prior to the i.v. injection of 3 MBq [18F]FDG per mouse. Static PET images were acquired 60 min p.i. using a β-cube PET scanner (Molecubes). The mice were kept under gas anesthesia (2.5% isoflurane in O2 at 1 L/min flow rate) during the entire procedure, with temperature and respiration were monitored throughout the scan. After the PET scanning, a CT image was acquired for anatomical co-registration using an X-cube CT scanner (Molecubes) as described above. The results have been included in the SI.
PET/MR imaging in a non-human primate
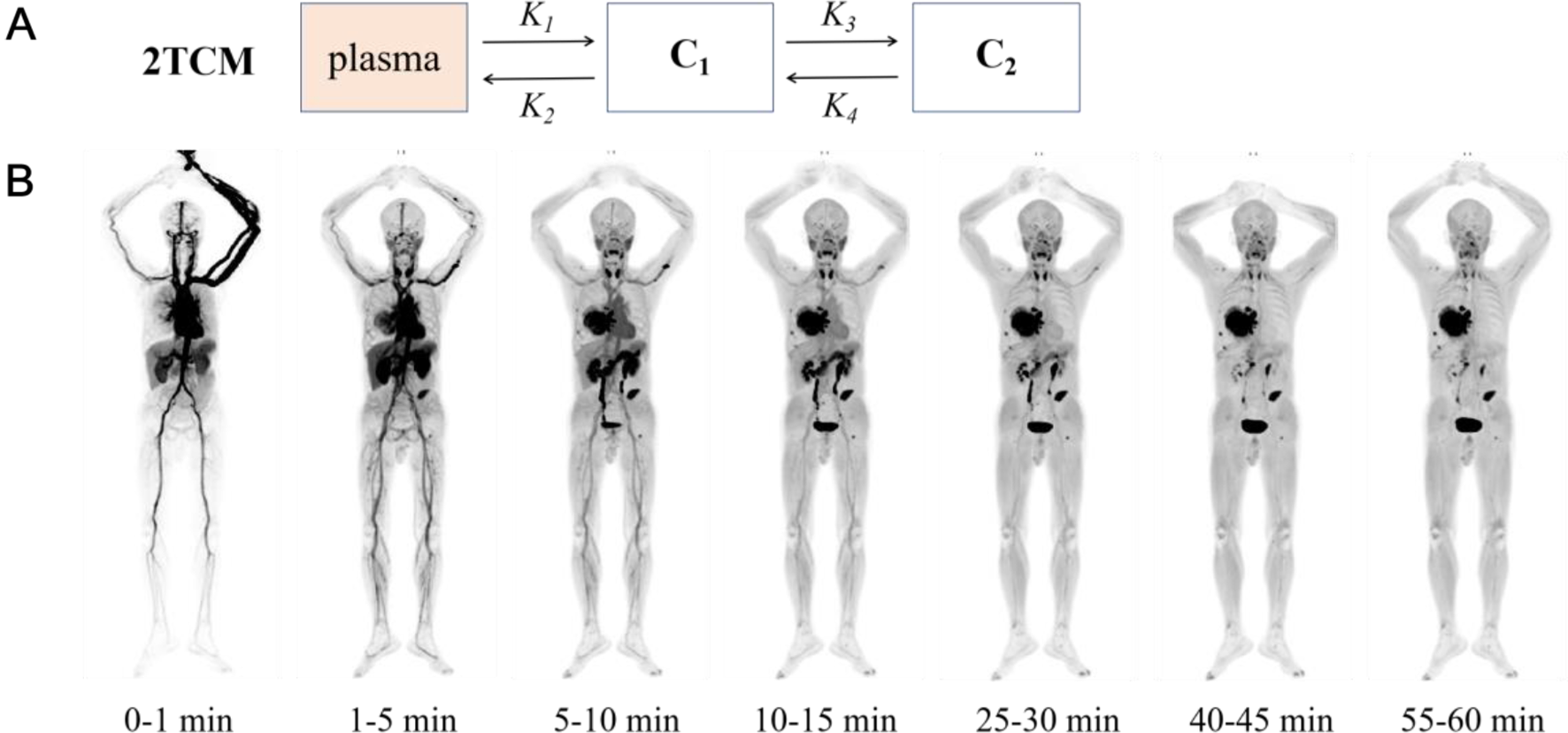
The non-human primate (Macaca mulatta, male, 9 years, 9.8 kg) was sedated with an intramuscular injection of 0.25 mL ketamine hydrochloride (100 mg/mL, Nimatek®) and 0.5 mL medetomidine hydrochloride (1 mg/mL, Domitor®) approximately 75 min prior to the initiation of PET scans, and transported to the PET facility. 60 min after the first injection, and subsequently every 30 min, the rhesus monkey received an additional dose of 0.125 mL ketamine hydrochloride and 0.25 mL medetomidine hydrochloride via i.v. injection. O2 saturation, CO2 level and heartbeat were constantly monitored. Temperature was regulated via a heating pad. Breathing frequency and eye response were checked regularly. 169 MBq (9.54 nmoles) [18F]AlF-NOTA-SC was injected i.v., and in vivo pharmacokinetics were determined by whole-body PET/ MR imaging of the tracer distribution up to 3 h after tracer injection. Blood samples were collected via venous blood sampling (vena saphena) at 10, 30 and 60 min p.i. to determine plasma metabolites using radioHPLC (procedure described below). After scanning (180 min p.i.), urine was collected by gently pressing the bladder, and urine metabolites were also determined by radioHPLC (procedure described below).
Image processing and analysis of naïve and tumor-bearing mice
PET data were histogrammed into 14 frames (4 × 15 s, 4 × 1 min, 1 × 5 min, 5 × 10 min) and reconstructed into a 192 × 192 image matrix with 0.4 mm voxels using 30 iterations the native Maximum-Likelihood Expectation-Maximization (MLEM) algorithm with corrections for randoms, scatter, attenuation and decay. CT data were reconstructed using a regularized statistical (iterative) image reconstruction algorithm with non-negative least squares, yielding an isotropic 200 μm voxel size. These data were scaled to Hounsfield Units (HUs) after calibration against a standard air/water phantom. Using PFUS v4.0 (PMOD Technologies GmbH, Zurich, Switzerland), fused PET-CT image were displayed, and volumes of interest were manually drawn over the organs or tumor of interest. Radiotracer uptake at 60 min, expressed as SUVmean, was the outcome measure.
Image processing and analysis of non-human primate
PET data were collected in list mode and corrected for randoms, scatter and deadtime. Attenuation correction was performed. Whole-body PET scans were acquired at 15, 32, 63, 91, 121 and 180 min post tracer injection. For each scan, a LAVA-Flex sequence (GE proprietary DIXON sequence, MP26) was acquired to generate the MR based attenuation map by measuring the in-phase (water and fat) and out-of-phase (water minus fat) T1-weighted signal. Water and fat tissues were segmented and assigned appropriate attenuation factors. In addition, a model-based lung segmentation was performed to assign the appropriate attenuation factor to lung tissue while for the head, the skull bone was included using an atlas-based approach. Ordered Subsets Expectation Maximization (28 subsets with 4 iterations), including time-of-flight information, resolution modeling, and Gaussian post smoothing with a Full-Width-at-Half-Maximum (FWHM) kernel of 4.5 mm (isotropic), was used for reconstruction.
All source organs with relevant detectable activity were delineated on the PET images with MR-AC map as guidance using PMOD version 4.1. Time-integrated activity coefficients (normalized cumulated activity; NCA) were calculated for each source organ by integrating their time-activity curves through curve fitting and normalizing the cumulated activity to the injected activity.
Image analysis was performed using PMOD version 4.1. VOIs were drawn over organs of interest on the PET image and SUVmean in these VOIs was determined.
Radiometabolite study in mice and rhesus macaque
The in vivo stability study was conducted following methods similar to those described Tshibangu et al. [38]. Briefly, naïve NMRI mice (n = 4) were anesthetized (2.5% isoflurane in O2 at 1 L/min flow rate) and injected via the tail vein with [18F]AlF-NOTA-SC (2–3 MBq). Urine samples were collected at 75 min p.i. by gently pushing the bladder, while plasma samples were collected at 10–75 min p.i. by collecting whole blood into K2EDTA MiniCollect tubes (0.5 mL Greiner Bio-One) after sacrificing the mice. Whole blood samples were counted in a gammacounter (Perkin-Elmer) followed by centrifugation at 2333 x g for 10 min to separate plasma, which was also counted in a gammacounter (Perkin-Elmer). Plasma proteins were precipitated by adding acetonitrile in a 1:1 ratio and filtration of the supernatant through a 0.22 μm filter (Millex-GV).
Similar procedures were applied to the non-human primate, with blood samples collected via the vena saphena at 10, 30 and 60 min p.i., and urine collection 180 min p.i. For analysis, 200 µL of plasma or urine samples were injected into the HPLC system described in the Radio-HPLC section. The retention time of the intact radiotracer was 18.1 min. The identity of the radiotracer was confirmed by comparison with a reference injection of the construct [natF]AlF-NOTA-SC, and by spiking blood samples of non-treated animals with the radiotracer. Plasma binding capacity was calculated by dividing the activity measured in plasma with the activity measured in the precipitated plasma proteins. Results are provided in the SI.
Statistical analysis
The results are reported as mean ± standard deviation (SD). Statistical significance was defined as p < 0.05 and was calculated using an independent samples t-test.









留言 (0)