Cell culture and treatment
Rat myocardial cell line H9c2 was provided by ATCC (Manassas, USA) and cultivated with culture medium consisted of DMEM medium (Gibco, USA), 10% FBS (Gibco), 100 µg/mL streptomycin and 100 U/mL penicillin obeying the manufacturer’s guidelines. These cells were cultured at 37 °C in the presence of 5% CO2.
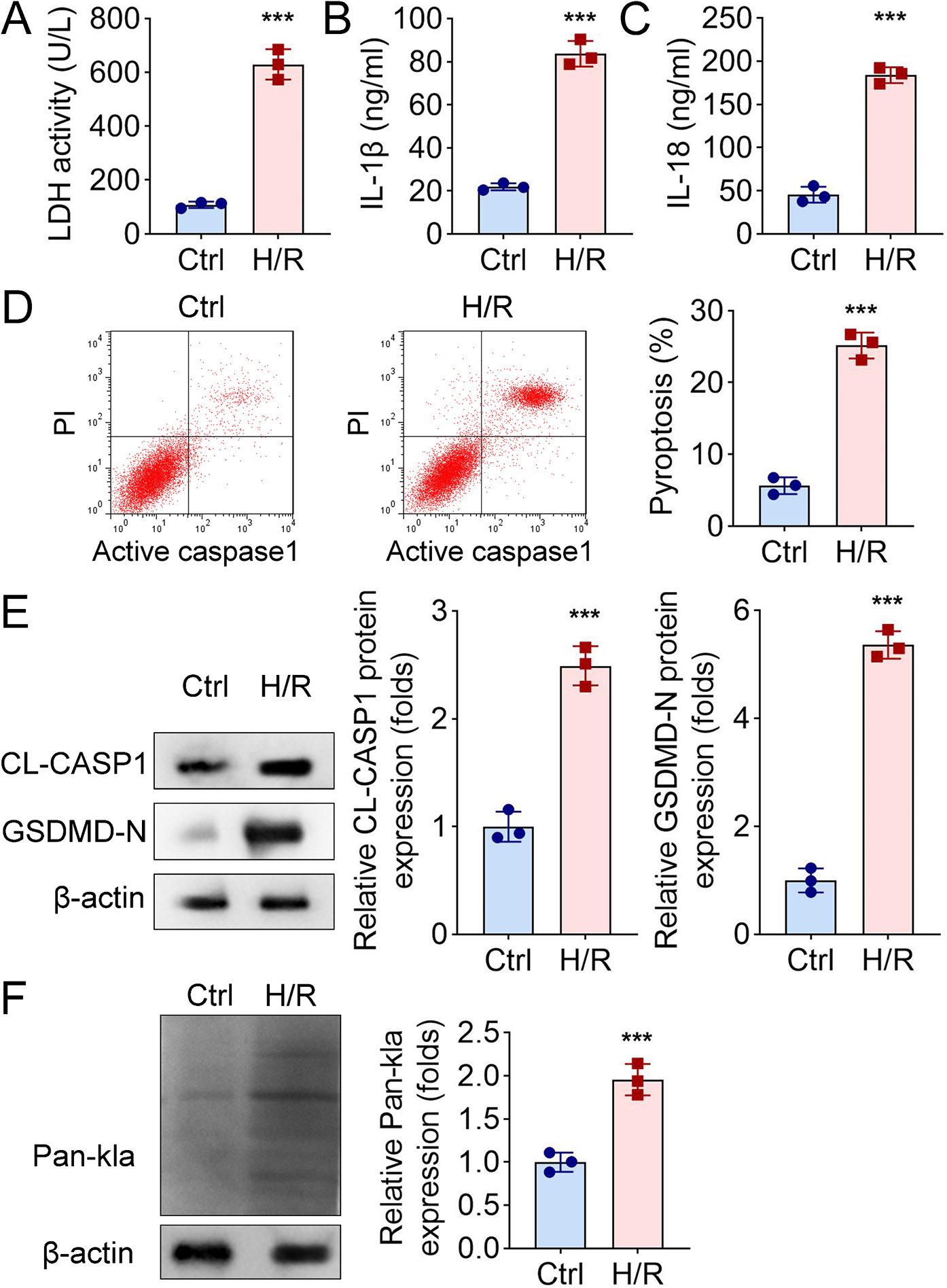
H9c2 cells were subjected to hypoxia/reoxygenation (H/R) to construct the cell model of I/R. For this purpose, H9c2 cells were maintained in the anoxic atmosphere of 94% N2, 5% CO2 and 1% O2 for 2 h and then reoxygenated under the environment of 95% air and 5% CO2 for additional 24 h. To upregulation of lactylation, cardiomyocytes were administered with 10 mM lactate for 6 d. In order to evaluate the protein stability of NLRP3, H9c2 cells were dealt with 100 µg/ml protein synthesis inhibitor cycloheximide (CHX, Sigma-Aldrich, USA). Besides, to activate NLRP3, the H9c2 cells were stimulated with the NLRP3 agonist, Nigericin (20 µM, Sigma-Aldrich), before H/R for 1 h.
Cell transfection
For silencing of LDHA, small interfering RNAs (siRNAs) specific for LDHA were acquired from RiboBio (Guangzhou, China). Non-specific siRNAs produced by RiboBio were employed as the negative control. Cell transfection was implemented by means of Lipofectamine® 2000 (Invitrogen, USA) abiding by the product protocols. H9c2 cells were collected at 24 h post transfection for H/R treatment. The used primers were listed below: siLDHA#1, 5’-CAAACTCCAAGCTGGTCATTA-3’, siLDHA2#, 5’-TGTTGATGTCATAGAAGATAA-3’, siNC, 5’-GAGATCCCGCTAACATCAAAT-3’.
CGTGTTATTGGAAGCGGTTG
Myocardial enzyme detection
To estimate the myocardial damage, the activity of myocardial enzymes was examined with colorimetric assay kits of lactate dehydrogenase (LDH) and creatine kinase-myocardial band (CK-MB), cardiac troponin I (cTnI) all obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). In line with instructions supplied by the vendor, cell or serum test samples were prepared and incubated with detection reagents. Absorbance was measured under a microplate reader (Varioskan LUX; Thermo Fisher Scientific, Waltham, MA, USA) the corresponding wavelength.
Enzyme-linked immunosorbent assay (ELISA)
Following the product manuals, the release of inflammatory cytokines interleukin (IL)-1β and IL-18 was determined by virtue of matched ELISA kits (R&D Systems, USA).
Flow cytometry
The pyroptosis of H9c2 cells was evaluated by flow cytometry analysis through the employment of a FAM-FLICA caspase-1 assay kit (ImmunoChemistry, USA). After different treatments, H9c2 cells were trypsinized, resuspended and treated with FAM-FLICA caspase-1 reagent for 1 h at 37 °C. Following rinsing thrice using washing buffer, cardiomyocytes were dyed by 100 µg/ml propidium iodide (PI) and then analyzed with a flow cytometer (Beckman Coulter, USA).
Western blot
H9c2 cells were treated with RIPA lysis buffer (Beyotime Biotechnology, China) to obtain total protein extracts. The BCA protein assay kit (Pierce, USA) was employed for quantification of protein concentration. Equivalent samples were detached by 10% SDS-PAGE and subsequently transferred onto PVDF membranes. After blockage with 5% defatted milk, membranes were immersed in primary antibodies at 4℃ throughout a night, followed by treatment with secondary antibodies (goat anti-rabbit; 1: 10,000, Abcam) at room temperature for 1–2 h and visualized by utilization of the chemiluminescence plus kit (Millipore, USA). The gray value of protein bands was analyzed with the ImageJ software. The following primary antibodies were adopted: anti-lysine lactylation (Kla) (1:1000, Micron Biotechnology Co., Ltd., China), anti-NLRP3 (1:1000, Abcam, UK), anti-ASC (1:1000, Abcam), anti-caspase-1 (1: 1000, Abcam), anti-cleaved caspase-1 (1:1000, Cell Signaling Technology, USA), anti-GSDMD-N (1: 1000, Biorbyt, UK), anti-LDHA (1:1000, Abcam), and anti-β-actin (1: 1000; Santa Cruz, USA). β-actin was used as the loading control.
Immunoprecipitation (IP)
The lactylation level of ASC, caspase-1, NLRP3, and GSDMD in H9c2 cells was assessed using an IP assay in combination with Western blot. In summary, cell lysates from H9c2 cells were collected and subjected to immunoprecipitation with ASC (1:30, Abcam), caspase-1 (1/50, Santa Cruz Biotechnology, Santa Cruz, CA, USA), NLRP3 (1:30, Abcam), and GSDMD (1:30, Abcam) and protein A/G agarose, followed by Western blot analysis targeting Kla.
Site mutation
To identify NLRP3 was lactylated at which site, we commissioned RiboBio Co. Ltd., to mutate lysine (K)245, K248, and K337 to arginine (K245R, K248R, and K337R). These mutated plasmids were then transfected into H9c2 cells.
Quantitative polymerase chain reaction (qPCR)
Total RNA from transfected H9c2 cells was extracted with Trizol reagent (Invitrogen). Following the directions of the First-Strand cDNA Synthesis Kit (Roche, Switzerland), reverse transcription was carried out. The SYBR qPCR Kit (Osaka, Japan) was applied to conduct the qPCR analysis of samples in the 7500 real-time PCR system (Applied Biosystems, USA). Relative gene expression was analyzed with the 2−ΔΔCT method and normalized to the endogenous reference β-actin. The sequences of primers were as follow: LDHA, 5’-CGTGTTATTGGAAGCGGTTG − 3’ (F) and 5’-TTCATTCCACTCCATACAGGC-3’ (R); β-actin, 5’-CTAAGGCCAACCGTGAAAAG-3’ (F) and 5’-ACCAGAGGCATACAGGGACA-3’ (R).
The mouse model of I/R
The procedures of animal experiments were performed under the permission of the Ethics Committee of MDKN Biotechnology Co., Lt. Twelve global LDHA knock-out (KO) C57BL/6J mice and 12 wild-type negative control of KO-LDHA mice were supplied by GemPharmatech (Nanjing, China). All mice were 6 weeks old and weighted 120 ± 10 g. The KO-NC mice were assigned to two group: the sham + KO-NC group and I/R + KO-NC group. Likewise, KO-LDHA mice were allocated into the I/R + KO-LDHA group and the I/R + KO-LDHA + nigericin group. To simulate myocardial I/R injury, mice were anesthetized by isoflurane via a rodent ventilator. The left anterior descending (LAD) coronary artery was exposed by incising the skin around the fourth intercostal space. Afterwards, LAD artery was subjected to 1 h of ligation followed by 1 h of reperfusion. Next, the wound was sewn up. The sham + KO-NC group mice underwent the same procedure, except for the absence of ligation. To determine the role of NLRP3 in myocardial I/R, the I/R + KO-LDHA + nigericin group mice were preoperatively administered with 5 mg/kg nigericin for a week. After plasma collection through retroorbital plexus hemorrhage, mice were euthanized.
2, 3, 5-triphenyltetrachloride (TTC) staining
TTC staining was implemented to assess the infarct area of cardiac tissues. After mice were sacrificed, 1% Evans Blue dye was infused into the aorta to determine the ischemic area. Subsequently, the heart tissues were preserved at -20℃ for 1.5 h, and then sliced into 2 mm Sect. 1% TTC solution was employed to stain tissue sections. The left ventricle (LV) was sectioned into five to seven transverse slices, and the analysis of the heart’s infarct size was conducted utilizing Image J software. Infarct area ratio = (sum of ischaemic area of each section)/ (sum of brain area of each section) × 100%.
Statistical analysis
GraphPad 5.0 software (San Diego, USA) was utilized for statistical analysis. Data were presented as the mean ± standard deviation (SD) from triplicate experiments. Comparison of two groups was completed with Student’s t-test. Differences between three or more groups were estimated by One-way ANOVA with Tukey’s post hoc analysis. Statistical significance was set as P < 0.05.








留言 (0)