
記住我







The Homo sapiens CERS6 gene, encoding the CerS6 protein (Uniprot Q6ZMG9), was cloned into the baculovirus transfer vector pHTBV1.1-CT10H-SIII-LIC (adapted from the BacMam vector pHTBV1.1; provided by F. Boyce, Massachusetts General Hospital) upstream of C-terminal tobacco etch virus (TEV)-cleavable 10xHis and Twin-Strep tags. The construct used for structural determination lacked the final 42 amino acids, which are predicted to be disordered. For in vitro biotinylation, the Avi-tagged CerS6 construct contained a Gly-Gly-Gly-Ser linker between CerS6 and the Avi tag, located upstream of the TEV cleavage site. Site-directed mutagenesis was performed using the Q5 site-directed mutagenesis kit (New England Biolabs).
Baculoviral DNA was generated by transposition of DH10Bac with the baculovirus transfer vector. Baculovirus was produced by transfecting Sf9 cells (Thermo Fisher Scientific) with the baculoviral DNA using the Insect GeneJuice transfection reagent (Merck Milipore). The virus was amplified by infecting Sf9 cells in the presence of 2% FBS and incubating for 72 h at 27 °C. For large-scale protein expression in Expi293F GnTI− cells (Thermo Fisher Scientific) in Freestyle 293 expression medium (Thermo Fisher Scientific), cells were transduced with baculovirus and supplemented with 5 mM sodium butyrate. Cells were incubated in a humidity-controlled orbital shaker at 37 °C with 8 % CO2 for 48 h and subsequently harvested by centrifugation at 900g for 15 min. Pelleted cells were washed with PBS, pelleted again by centrifugation, flash-frozen in liquid N2 and stored at −80 °C. To compare mutant activity, WT and mutant proteins were expressed in Expi293F cells (Thermo Fisher Scientific) using the same protocol.
Large-scale CerS6 purificationCerS6-overexpressing cells were resuspended in buffer A (50 mM HEPES pH 7.5, 200 mM NaCl and 5% (v/v) glycerol) and solubilized with 1% (w/v) lauryl maltose neopentyl glycol (LMNG) (Anatrace) and 0.1% (w/v) cholesteryl hemisuccinate (CHS) (Sigma-Aldrich). Insoluble material was removed by centrifugation at 35,000g and the protein was purified from the supernatant by batch binding to Strep-Tactin XT Superflow resin (IBA Lifesciences). The resin was washed initially with buffer A containing 0.05% (w/v) GDN and subsequently with buffer A supplemented with 1 mM adenosine triphosphate (ATP; Sigma-Aldrich), 10 mM MgCl2 and 0.05 % (w/v) GDN. CerS6 was eluted from the resin using buffer A supplemented with 100 mM d-biotin (Fluorochem) and 0.02% (w/v) GDN, while the C-terminal tag was cleaved by TEV protease digestion overnight. The His-tagged TEV protease was removed by binding to Co2+-charged TALON resin (Clontech) and the flowthrough was concentrated using a concentrator with a 100-kDa molecular weight cutoff (Corning). The protein was finally purified by SEC on a Superdex 200 Increase 10/300 GL column (GE Healthcare), pre-equilibrated in SEC buffer (20 mM HEPES pH 7.5, 200 mM NaCl and 0.01 % (w/v) GDN).
For nanobody generation and selection, GDN was replaced in the purification buffers with 0.003% (w/v) LMNG and 0.0003% (w/v) CHS. For in vitro biotinylation of C-terminally Avi-tagged CerS6, during the TEV cleavage step, the protein sample was further supplemented with 15 mM MgCl2, 15 mM ATP, 50 mM bicine pH 8.3 and BirA at a 1:15 (w/w) BirA:CerS ratio. Biotinylation efficiency was routinely monitored by denaturing intact protein MS.
For structural determination, CerS6 was mixed with a 1.5× molar excess of nanobody (either Nb22 or Nb02) and incubated for 1 h on ice. The CerS6–nanobody complexes were purified by SEC, concentrated to 5 mg ml−1 using a concentrator with a 100-kDa molecular weight cutoff (Sartorius) and processed immediately for cryo-EM.
Nanobody library generation and selectionPurified CerS6 was reconstituted at a 1:20 (protein:lipid (w/w) ratio into liposomes composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol, 1,2-dimyristoyl-sn-glycero-3-phosphocholine and 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (7:3:7:3 (w/w)) as previously described53.
To obtain anti-CerS6 nanobodies, alpacas were immunized and the nanobody library was generated as previously described54, with the exception that 200 µg of purified CerS6 in proteoliposomes was used for each immunization. All the procedures concerning alpaca immunization were approved by the Cantonal Veterinary Office of Zurich (license no. ZH 198/17). The resulting nanobody library was screened by biopanning against CerS6. Subsequently, 190 single clones from the enriched nanobody library were analyzed by ELISA for binding to CerS6. A total of 96 ELISA-positive clones were Sanger-sequenced and grouped in families according to their complementarity-determining region length and sequence diversity55. Of these, 42 unique nanobodies were identified as belonging to 26 nanobody families and were taken for further validation.
Nanobodies were expressed in 50-ml scale in WK6 cells and purified from periplasmic extracts using Ni2+-NTA resin, as previously described55. Unique nanobodies were screened by biolayer interferometry (BLI) in an Octet Red 384 system (Sartorius) using Streptavidin SA biosensors (Sartorius) loaded with 40 µg ml−1 biotinylated CerS6 in SEC buffer containing 0.003% (w/v) LMNG and 0.0003% (w/v) CHS. Nanobodies with slow off rates were identified and nanobodies Nb22 and Nb02 were prioritized for structural studies.
Thermal stability measurementsThermal unfolding experiments were carried out by measuring intrinsic tryptophan fluorescence on a Prometheus NT.48 instrument (NanoTemper Technologies) using 5 µM CerS6 solubilized in 0.003% LMNG and 0.0003% CHS. The protein was incubated for 1 h at 4 °C in the presence or absence of 20 or 100 µM FTY720 (Sigma-Aldrich) or FB1 (Sigma-Aldrich). Proteins were heated from 20 °C to 95 °C, at a rate of 1 °C min−1, and unfolding was monitored by the ratio of fluorescence emission at 350 nm and 330 nm. The melting temperature was determined from the inflection point of the transition using the PR.ThermControl software (NanoTemper Technologies).
Cryo-EM sample preparation and data acquisitionThe purified CerS6–Nb22 and CerS6–Nb02 complexes (5 mg ml−1) were applied to freshly glow-discharged Quantifoil 200-mesh Au R1.2/1.3 grids. Plunge-freezing in liquid ethane was carried out using a Vitrobot Mark IV (Thermo Fisher Scientific) set to 4 °C and 100 % humidity. For the CerS6–Nb22–FB1 complex, the SEC-purified CerS6–Nb22 complex was incubated with 120 µM FB1 (Sigma-Aldrich) for 10 min on ice before protein concentration and subsequently for an additional 2 h on ice before grid preparation. EM grids were screened on a Glacios (Oxford Particle Imaging Center (OPIC)) and high-resolution data were collected on a Titan Krios G3 microscope (Leicester Institute of Structural and Chemical Biology (LISCB)) operating at 300 kV and equipped with a Bioquantum energy filter (Gatan) (operated at 20-eV slit width) and a K3 direct electron detector (Gatan) at ×130,000 nominal magnification, in super-resolution mode (2× binning; physical pixel size: 0.656 Å per pixel), using a defocus range between −0.8 µm and −2.4 µm. The total exposure dose was 56.3 e− per Å2, fractionated over 50 frames. In total, 14,309, 18,386 and 14,656 videos were collected for the CerS6–Nb22, CerS6–Nb22–FB1 and CerS6–Nb02 datasets, respectively.
Cryo-EM data processing and model buildingMovies were motion-corrected using MotionCor2 (ref. 56) and contrast transfer function (CTF) estimation was carried out in cryoSPARC57 (version 3.3.1). Micrographs with bad CTF fitting (>5 Å) were excluded, yielding 14,196, 16,647 and 14,596 micrographs for further analysis for the CerS6–Nb22, CerS6–Nb22–FB1 and CerS6–Nb02 datasets, respectively.
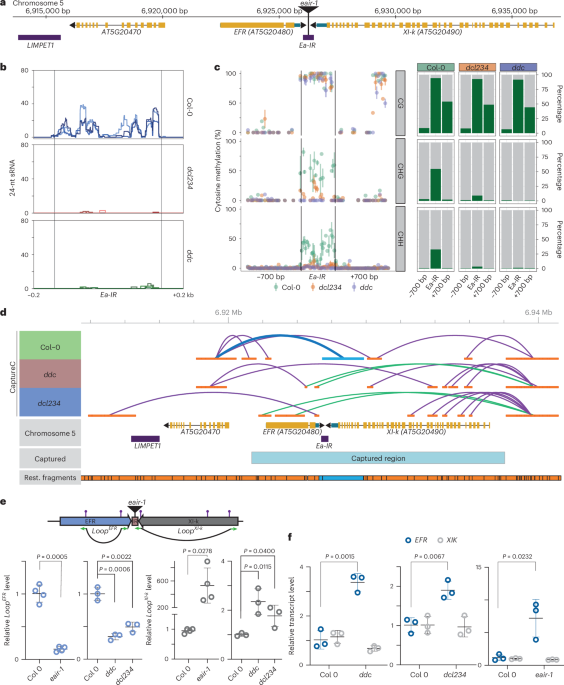
For the CerS6–Nb22 covalent acyl–enzyme intermediate dataset, particles were initially blob-picked and extracted from a subset of micrographs; then, representative well-resolved two-dimensional (2D) classes were used for template-based picking of the entire dataset. A total of 3,400,118 template-picked particles were extracted in a 360-pixel box Fourier-cropped to 120 pixels, corresponding to a pixel size of 1.968 Å per pixel, and classified in two rounds of reference-free 2D classification yielding 1,261,928 good particles. Four ab initio models were generated and used as reference models in heterogeneous refinement. Particles belonging to the well-resolved 2:2 CerS6–Nb22 dimer complex class (53% of input particles) were selected and subjected to a further round of heterogeneous refinement. The best-resolved particles (497,504) were then subjected to nonuniform refinement58 with C2 symmetry imposed, yielding a 3.21-Å reconstruction (Fourier shell correlation (FSC) = 0.143). These aligned particles were imported into RELION using the csparc2star.py script from the University of California, San Francisco pyem package59 and re-extracted in a 360-pixel box Fourier-cropped to 270 pixels, corresponding to a pixel size of 0.875 Å per pixel. In RELION, CTF parameters were refined, followed by Bayesian particle polishing60. The polished particles were subjected to three-dimensional (3D) classification without image alignment (k = 10, T = 12). Certain classes displayed worse side-chain density and local deviations from C2 symmetry. Therefore, to improve map quality, the 93,680 particles belonging to the two highest-resolution C2-symmetric classes were reimported into cryoSPARC and subjected to a final nonuniform refinement with C2 symmetry, yielding a 3.22-Å reconstruction (FSC = 0.143). Importantly, even though CTF refinement, Bayesian polishing and 3D classification in RELION did not improve the nominal resolution, the final reconstruction revealed better-defined features, including improved side-chain density in the CerS6 active site (Extended Data Fig. 2f,g).
The CerS6–Nb22 N-acyl FB1 dataset was processed using the same workflow as the covalent acyl–enzyme intermediate dataset, with some changes. Briefly, 3,998,935 template-picked particles were classified in two rounds of reference-free 2D classification, yielding 1,353,329 good particles. Four ab initio models were generated and used as reference models in heterogeneous refinement. A total of 641,462 particles belonging to the well-resolved 2:2 CerS6–Nb22 dimer complex class (48% of input particles) were subjected to nonuniform refinement with C2 symmetry imposed, yielding a 3.17-Å reconstruction. After CTF refinement, Bayesian polishing and 3D classification without image alignment in RELION, the 154,239 particles belonging to the three highest-resolution C2-symmetric classes were selected. These particles were reimported into cryoSPARC and subjected to a final nonuniform refinement with C2 symmetry, yielding a 2.95-Å reconstruction (FSC = 0.143).
The CerS6–Nb02 covalent acyl–enzyme intermediate dataset was processed similarly to the other datasets, with some modifications. Briefly, 4,497,470 template-picked particles underwent two rounds of reference-free 2D classification, leading to the identification of 1,025,813 good particles. Following heterogenous refinement, 507,128 particles belonging to the well-resolved 2:2 CerS6–Nb02 dimer complex class (50% of input particles) were subjected to nonuniform refinement with C2 symmetry imposed, yielding a 3.22-Å reconstruction. Particles were imported into RELION and re-extracted in a 432-pixel box Fourier-cropped to 324 pixels, corresponding to a pixel size of 0.875 Å per pixel. After CTF refinement, Bayesian polishing and 3D classification without alignment in RELION, the 153,485 particles belonging to the two highest-resolution C2-symmetric classes were selected. These particles were then reimported into cryoSPARC for a final nonuniform refinement with C2 symmetry applied, yielding a 3.02-Å reconstruction (FSC = 0.143).
Atomic models were generated by fitting the AF2 (ref. 61) prediction of CerS6 (AF-Q6ZMG9-F1) and Phyre2 (ref. 62) nanobody homology models into the cryo-EM maps and subsequently manually adjusted in Coot63. In all datasets, residues 72–119, corresponding to the Hox-like domains, were poorly resolved in the sharpened maps. However, their position was evident in the unsharpened and blurred maps (Extended Data Figs. 2e and 3a). To model this region, we used tight restraints to the AF2 prediction for this domain and docked it into the envelope of the blurred maps (Bblur = 200 Å2) and surface-exposed side chains were truncated at Cβ. The atomic models were refined using PHENIX real-space refinement64 with secondary structure and Ramachandran restraints and noncrystallographic symmetry constraints. Restraint dictionaries for FB1 and the covalently attached C16:0 chain were generated using AceDRG65. The final models comprised residues 2–330 (CerS6–Nb22 and CerS6–Nb02 covalent intermediate state) or 2–334 (CerS6–Nb22 N-palmitoyl FB1-bound state) of CerS6, residues 1–124 of Nb22 or residues 1–123 of Nb02, the C16:0 chain covalently attached to His211 of CerS6 or the N-palmitoyl FB1 product, one POPC molecule and the first N-acetylglucosamine (GlcNac) residue of the N-linked glycan visible on Asn18. Weighted Fo − Fc ligand difference maps were calculated using Servalcat66, available in CCPEM67, by omitting the ligands from the atomic models. All descriptions and figures are based on the structures of the CerS6–Nb22 complex unless otherwise stated. Structural similarity to other acyl-CoA-binding proteins was identified using DALI68.
Denaturing intact protein MSAll MS experiments were carried out on purified CerS6 in the absence of nanobodies. The intact masses of purified protein samples were analyzed by denaturing intact protein MS, conducted using an Agilent 1290 Infinity liquid chromatography (LC) system in line with an Agilent 6530 accurate-mass quadrupole time-of-flight MS instrument (Agilent Technologies), as previously described33. Typically, 5–8 μg of purified protein (at 1.5–2.0 mg ml−1), diluted to 20 μl in 30% methanol in 0.1% formic acid, was used per injection. Data were acquired between 100 and 3,200 m/z and analyzed using MassHunter Qualitative Analysis version B.07.00 (Agilent Technologies) software. Peaks between 650 and 3,200 m/z in the sum of the mass spectra obtained during protein elution were deconvoluted using the maximum entropy charge deconvolution algorithm.
The identity of the deconvoluted mass peaks was assigned initially on the basis of the expected mass of the purified proteins and subsequently the observed mass shifts between peaks in the deconvoluted mass spectra. The lower-molecular-weight CerS6 peak corresponded to loss of the initiator methionine (theoretical mass shift of −131.20 Da), acetylation of the new N terminus (theoretical mass shift of +42.04 Da) and addition of an N-linked GlcNAc (theoretical mass shift of +203.19 Da). However, the major glycosylation species observed contained the complete core N-linked glycan (theoretical mass shift of +1217.05 Da), as expected for protein expressed in Expi293F GnTI− cells. In addition, we observed mass shifts of +237.47 and +238.51 Da relative to these two glycosylated species, respectively, which we interpreted as palmitoylation at a single site (theoretical mass shift of +238.41 Da), corresponding to the covalent acyl–imidazole intermediate observed in the cryo-EM structure. One additional modification was observed (approximately +264 Da) but its identity could not be assigned. This unknown modification occurred at a distinct site to that of the palmitoylation, as both modifications could occur simultaneously in the same protein molecule (Extended Data Fig. 1f,g), thus excluding the possibility of this unknown modification occurring in the active site.
To monitor the reaction of the covalent intermediate species with the second substrates, before denaturing intact protein MS analysis, the protein (2 mg ml−1) was incubated with 200 µM sphinganine (Avanti Polar Lipids), 200 µM FB1 (Sigma-Aldrich) or 600 µM FTY720 (Sigma-Aldrich) for 90 min at 37 °C. All intact mass experiments were conducted at least twice using distinct biological samples. Replicate deconvoluted mass spectra are shown in Extended Data Fig. 6.
Product detection by LC–HRMSProduct detection by LC–HRMS was conducted on a nanoElute LC system in line with a timsTOF Pro 2 MS instrument (Bruker). The reactions were set up as described for the denaturing intact protein MS experiments, diluted 1:40 (v/v) in 30% methanol in 0.1% formic acid, and 1 µl of each sample was injected onto an IonOpticks C18 nano ultrahigh-performance LC column (1.6-μm particle size; 0.075 mm × 250 mm).
The flow rate was set to 0.5 µl min−1 and the solvent system consisted of 0.1% Optima LC–MS-grade formic acid (Fisher Chemical) in high-performance LC electrochemical-grade water (Fisher Chemical) (solvent A) and 0.1% formic acid in Optima LC–MS-grade methanol (Fisher Chemical) (solvent B). The initial condition was 60% solvent B and a linear gradient from 60% to 95% solvent B was applied over 17.8 min to elute the samples. This was then followed by a final 2.2-min isocratic elution with 95% solvent B before the system was re-equilibrated between samples for 5 min with 60% solvent B.
The MS instrument was operated in positive ion mode with a capillary voltage of 1,600 V and the drying gas was supplied at 180 °C with a flow rate of 3 L min−1. Additional parameters were as follows: deflection delta, 70 eV; funnel 1 radiofrequency (RF), 350 Vpp; funnel 2 RF, 600 Vpp; multipole RF, 500 Vpp. Data were acquired between 150 and 2,200 m/z and analyzed using the Bruker Compass DataAnalysis 5.3.556 software. The extracted ion chromatograms (EICs) are presented in Figs. 3c and 4d and correspond to the theoretical [M + H]+ ions (tolerance: ±0.005 m/z) of the reaction products N-palmitoyl dihydrosphingosine (C16:0 ceramide) (theoretical m/z: 540.5350), N-palmitoyl FB1 (theoretical m/z: 960.6254) and N-palmitoyl FTY720 (theoretical m/z: 546.4881).
LC–electrospray ionization (ESI)-MS/MS characterization of N-palmitoyl FTY720To structurally characterize the proposed N-palmitoyl FTY720 reaction product, purified protein was incubated with 600 μM FTY720 as before and the reaction mixture was initially analyzed by LC–ESI-MS on an Agilent 1290 Infinity LC system in line with an Agilent 6530 accurate-mass quadrupole time-of-flight MS instrument (Agilent Technologies) as described above. This enabled the identification of the putative [M + H]+ ion of the N-palmitoyl FTY720 product (observed m/z: 546.4849; theoretical m/z: 546.4881). Its product ion spectrum was then obtained by LC–ESI-MS/MS. For this purpose, the MS instrument was operated in positive ESI mode (4 GHz). MS parameters were as follows: capillary voltage, 4,000 V; fragmentor voltage, 175 V. Data were acquired between 100 and 1,700 m/z. The targeted parent ion (546.4881 m/z; retention time, 8.713 min) was fragmented using a collision energy of 14 V.
Dihydroceramide synthase activity measurementsFor activity assays, WT or mutant CerS6 proteins were overexpressed in Expi293F cells and membranes were prepared as follows. The cell pellet from 0.5 L of culture was thawed in PBS and lysed using an Emulsiflex C5 homogenizer (Avestin); then, cell debris was removed by centrifugation. Membranes were subsequently isolated by ultracentrifugation at 160,000g for 90 min, resuspended in assay buffer (20 mM HEPES pH 7.5, 25 mM KCl, 1 mM MgSO4 and 0.1% (v/v) glycerol), flash-frozen in liquid N2 and stored at −80 °C.
On the day of the assay, membranes were thawed and diluted to 0.25 mg ml−1. Next, 20 µl of diluted membranes were dispensed per well on flat-bottom polystyrene 384-well Lumitrac plates (Greiner Bio One). Then, 50 µM sphinganine (2.5 µl of 500 µM sphinganine in assay buffer containing 10% ethanol) and 50 µM palmitoyl-CoA (Sigma-Aldrich) (2.5 µl of 500 µM palmitoyl-CoA in assay buffer) were added to each well for a final reaction volume of 25 µl. For untreated controls, 5 µl of assay buffer were added instead of the substrates. The plates were then incubated for 1 h at room temperature and the reaction was terminated by the addition of 40 µl of butanol spiked with N-palmitoyl(d9) dihydrosphingosine (Avanti Polar Lipids) to yield a final concentration of 5 µM N-palmitoyl(d9) dihydrosphingosine as an internal standard. The plates were then shaken at 1,800 rpm for 2 min and centrifuged at 1,000g for 30 s. Finally, 40 µl of the organic (upper) phase was transferred into 384-well polypropylene deep-well plates (Greiner Bio One) and diluted with 40 µl of butanol.
The analytical sample handling was performed by a rapid-injecting RapidFire autosampler system (Agilent) coupled to a triple-quadrupole MS instrument (Triple Quad 6500, AB Sciex) as previously described69, with some modifications. Briefly, the liquid sample was aspirated by a vacuum pump into a 10-µl sample loop for 6,000 ms and subsequently flushed for 3,000 ms onto a C4 cartridge (Agilent) with the aqueous mobile phase (99.5% water, 0.49% acetic acid and 0.01% trifluoroacetic acid; flow rate, 1.5 ml min−1). The multiple reaction monitoring transition for C16:0 dihydroceramide is 540.5 → 266.3 m/z (declustering potential, 130 V; collision energy, 38 V) and that for the internal standard C16:0 (d9)dihydroceramide is 549.5 → 266.3 m/z (declustering potential, 130 V; collision energy, 38 V). The MS instrument was operated in positive ion mode (curtain gas, 35 arbitrary units (AU); collision gas, medium; ion spray voltage, 4,200 V; temperature, 550 °C; ion source gas 1, 65 AU; ion source gas 2, 80 AU). MS data processing was performed in Gubbs Mass Spec Utilities and peak area ratios between C16:0 dihydroceramide and the internal standard were calculated.
Expression of each mutant in the membrane samples was evaluated through western blotting using 10 ng ml−1 Strep-Tactin conjugated with horseradish peroxidase (IBA Lifesciences), applying SYPRO ruby (Thermo Fisher Scientific) staining as a loading control (Fig. 3e). Band intensity was quantified using the GeneTools software (Syngene) and activity was normalized to protein expression.
MD simulationsAtomistic MD simulations were performed using the Desmond software package (D. E. Shaw Research) within the Maestro software suite (Schrödinger). The simulation setup and analysis were carried out as follows. For each of the two states, a CerS6 monomer was embedded in a lipid bilayer consisting of 99 POPC molecules after undergoing the protein preparation step as implemented in Maestro. The dimensions of the simulation cell were approximately 90 × 70 × 70 Å, with a minimum distance of 10 Å between the protein and the cell boundaries. The systems contained 8,855 (covalent intermediate bound state) or 9,368 (N-acyl FB1-bound state) water molecules and eight or six corresponding chloride counterions to maintain overall charge neutrality. The simulations were performed using the OPLS4 force field70 for the protein and lipid molecules and the simple point-charge water model for the water molecules. The system was set up using the Maestro software suite (Schrödinger). The simulations were run in Desmond using an NPT ensemble at a temperature of 300 K. A timestep of 2 fs was used for the integration of equations of motion. Four independent simulations were performed, each with a duration of 100 ns, resulting in a total simulation time of 400 ns. Snapshots of the system were recorded every 100 ps for further analysis.
FiguresFigures depicting molecular models were generated using PyMOL (Schrödinger) and ChimeraX71.
Reporting summaryFurther information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
留言 (0)