
記住我
The human immune system is a vital defense mechanism that comprises immune organs, cells and molecules, playing a crucial role in identifying and eliminating invading pathogens while maintaining the body’s internal environment stability and physiological balance. However, when the immune system becomes dysregulated, it can generate responses against self-antigens, leading to the attack of normal tissue cells and development of autoimmune diseases (AIDs) (1, 2). AIDs encompass a broad range of disorders triggered by factors such as the appearance of self-antigens, immune dysregulation, cross-reactivity, and genetic predisposition. Clinical AIDs mainly include rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), multiple sclerosis (MS), dermatomyositis (DM) and other diseases (1–3). The onset and progression of AIDs are closely associated with this prominent hypoxic microenvironment (4, 5). Notably, hypoxia-dependent and non-hypoxia-stimulated regulatory pathways mediated by hypoxia-inducible factor-1 (HIF-1) in the hypoxic microenvironment play a pivotal regulatory role in the pathogenesis of AIDs (6, 7). The Hypoxic Microenvironment can be defined as a pathophysiological state in which the concentration of oxygen in a particular tissue area is significantly reduced (8). The formation of this microenvironment is the result of the interaction of multiple factors. As target tissues undergo destruction, a large number of immune cells, including macrophages, neutrophils, myeloid-derived suppressor cells (MDSCs), and T and B lymphocytes, infiltrate and become activated. This infiltration contributes to the formation of a hypoxic microenvironment. The ongoing inflammatory reactions and aberrant immune activation in this microenvironment lead to increased oxygen consumption, further exacerbating local tissue hypoxia (4, 5, 9). Conversely, local tissue hypoxia intensifies inflammatory reactions and immune dysfunction, further exacerbating the imbalance between oxygen supply and demand in local areas. Hypoxia, characterized by an imbalance between oxygen supply and consumption within cells, is an important feature of AIDs (10, 11). In patients with AID, under hypoxic microenvironment conditions, immune cells undergo excessive activation, abnormal blood vessel proliferation, metabolic reprogramming, and deposition of immune complexes, disrupting self-recognition, interfering with immune tolerance, and promoting the formation of autoantibodies, thus triggering immune reactions between autoantibodies or sensitized lymphocytes and self-tissue antigens (12, 13). Moreover, factors such as adaptability to hypoxic environments, drug resistance, and immune evasion are crucial determinants affecting the progression and prognosis of AIDs (14–17). Therefore, in this review, we summarize the impact of the hypoxic microenvironment on AIDs and elucidate relevant molecular mechanisms, providing guidance for further exploration of the molecular mechanisms underlying the occurrence and development of AIDs in hypoxic environments.
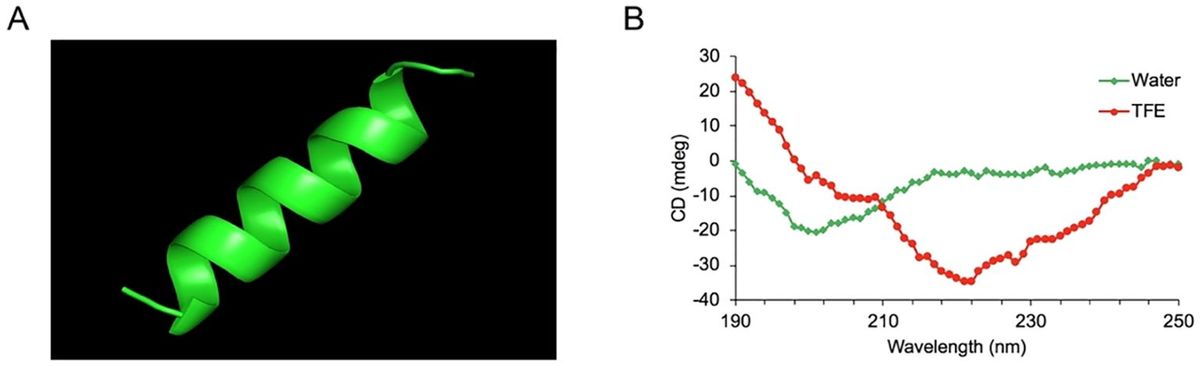
2 Overview of hypoxic microenvironment and hypoxia-inducible factorsIn 1992, Semenza et al. discovered the transcription factor HIF associated with hypoxic stress under low-oxygen conditions (18). Subsequently, Ratcliffe et al. confirmed HIF’s involvement in cellular glycolysis, demonstrating that under hypoxic conditions, the expression levels of phosphoglycerate kinase (PGK) and lactate dehydrogenase A (LDHA) are significantly upregulated (19). Kaelin et al. elucidated that the tumor suppressor protein Von Hippel-Lindau Tumor Suppressor Protein (pVHL) negatively regulates HIF, and loss of pVHL hinders HIF degradation, thereby promoting tumorigenesis (20, 21). Forsythe et al. confirmed that the upregulation of HIF-1α facilitates the expression of vascular endothelial growth factor (VEGF), whereas the absence of HIF-1α inhibits angiogenesis, resulting in embryonic death (22, 23). In recent years, the impact of hypoxic microenvironments on HIF expression and its multifaceted involvement in cellular processes, including metabolism, proliferation, migration, and activation, has garnered considerable attention from researchers, rendering it a prominent and focal area of investigation. Furthermore, its important role in various diseases, such as cancer, autoimmune disorders, and microbial infections, underscores its significance in biomedical research (Figure 1).
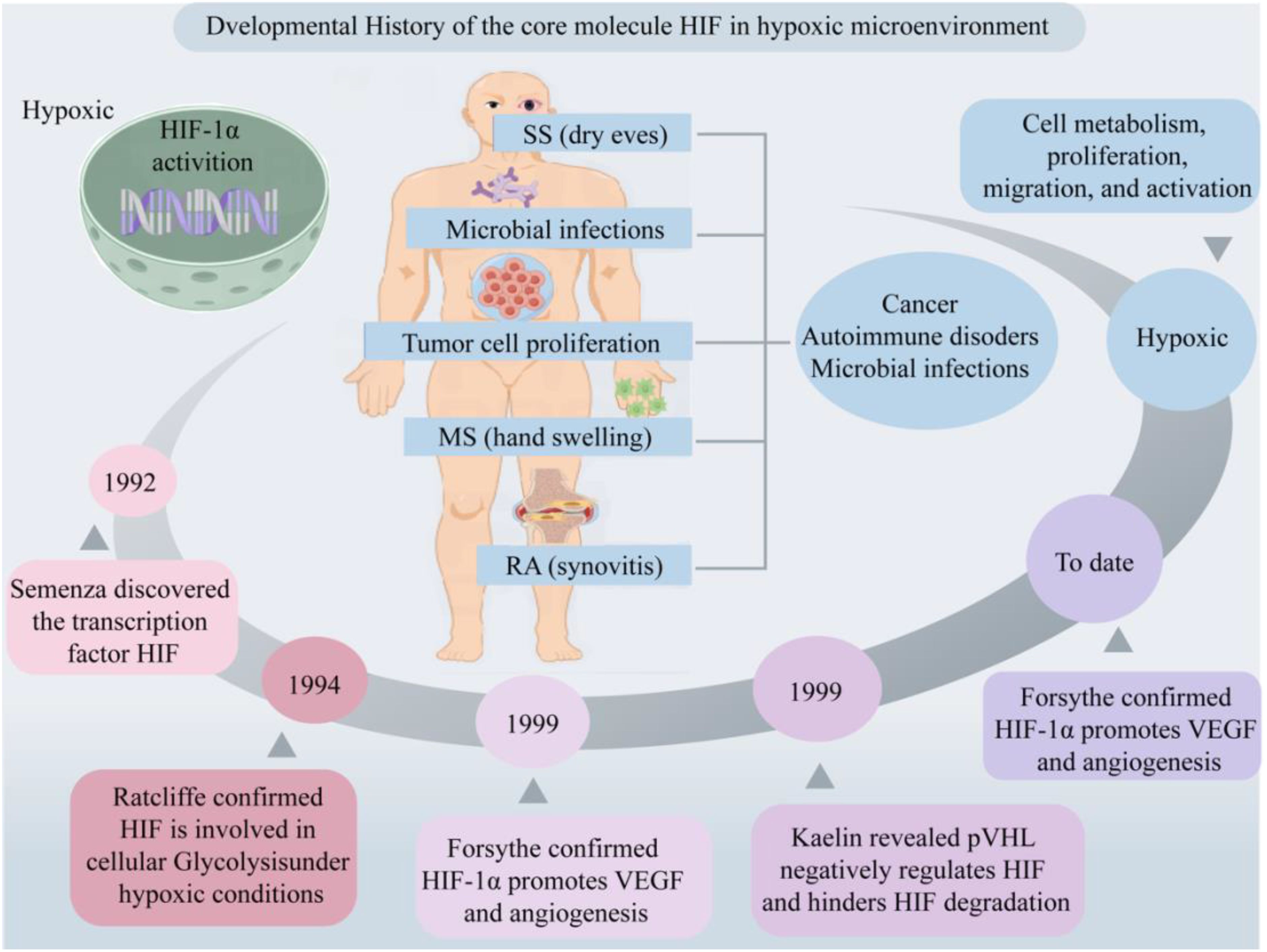
Figure 1. Developmental history of the core molecule HIF in hypoxic microenvironment. Hypoxic microenvironments are involved in cell metabolism, proliferation, migration and activation, mediating the occurrence of diseases such as cancer, AIDs and microbial infections. Created with figdraw.com.
2.1 Structure of HIF in hypoxic microenvironmentIn hypoxic conditions, HIF serves as the primary signaling molecule, with HIF-1α exhibiting high sensitivity to low oxygen and serving as the core regulatory molecule in the body’s response to hypoxia (6). HIF is a heterodimer composed of an oxygen-sensitive α subunit and a constitutively expressed β subunit. The α subunit comprises three isoforms: HIF-1α, HIF-2α, and HIF-3α, which are regulated by oxygen. It contains an oxygen-dependent degradation domain (ODD) crucial for HIF degradation regulation (4). Under normoxic conditions (21% O2) HIF exhibits some expression, yet the synthesized proteins undergo rapid ubiquitination and subsequent degradation via the oxygen sensor prolyl hydroxylases (PHDs)/HIF-1α/pVHL pathway (24). Only under hypoxic conditions can HIF-1 achieve stable expression (25). Reports suggest that HIF-1α is upregulated under intense hypoxia or early hypoxia (<24 hours), while HIF-2α and HIF-3α are highly expressed under mild or chronic hypoxia (>24 hours) (26, 27). Moreover, another study found that HIF-1α is widely expressed and regulates oxygen homeostasis most significantly in mammals, whereas HIF-2α exhibits strong tissue specificity, with its functions overlapping or differing from those of HIF-1α (28). The β subunit includes HIF-1β, which can be expressed in the cytoplasm or nucleus, unaffected by oxygen concentration, playing a role in maintaining HIF conformation and stable expression (4). Collectively, this indicates that different subunits of HIF have slightly different expression patterns and functional roles in the hypoxic microenvironment.
2.2 Function of HIF in hypoxic microenvironmentUnder normoxia conditions, the hydroxylation of Pro402 and Pro564 residues within the ODD domain of HIF-1α, catalyzed by oxygen, iron, and 2-oxoglutarate, results in conformational alterations that bolster its affinity for PHD enzyme recognition. This, in turn, initiates the subsequent association of HIF-1α with pVHL, leading to the formation of a stable HIF-1α/pVHL complex (29). This complex subsequently undergoes ubiquitination by an array of ubiquitin proteins (Ub), ultimately culminating in the proteasomal degradation of HIF-1α under normoxic conditions (30, 31) (Figure 2A). Under hypoxic conditions, the functionality of HIF is primarily mediated by the α subunit, where intracellular oxygen concentration affects the protein stability and transcriptional activity of HIF-1α (6). The upregulation of reactive oxygen species (ROS) expression inhibits the enzymatic activity of PHDs and factor-inhibiting HIF (FIH), subsequently resulting in the accumulation of HIF-1α within the cytoplasm (32, 33). FIH hydroxylates the transactivation domain (TAD) of HIF-1α at the C terminus, where the asparagine residue Asn803 significantly reduces the transcriptional activity of HIF-1α (34). During this process, cytoplasmic HIF-1α translocations to the nucleus, where it integrates with HIF-1β and cofactor proteins p300/CBP to form a heterodimer, thereby rendering it less vulnerable to proteasomal degradation. This heterodimer subsequently recognizes and binds to hypoxic response elements (HREs), thereby activating downstream gene transcription (4). This, in turn, promotes glucose uptake and metabolism, upregulates glycolysis via the activation of glucose transporter 1 (GLUT1) (35), stimulates erythropoiesis through the upregulation of erythropoietin (EPO) essential for red blood cell production, and mediates vascular angiogenesis via VEGF (22, 36). This illustrates that HIF activates a series of adaptive responses in hypoxic microenvironments by regulating its stability and transcriptional activity to maintain cell and tissue function and survival.
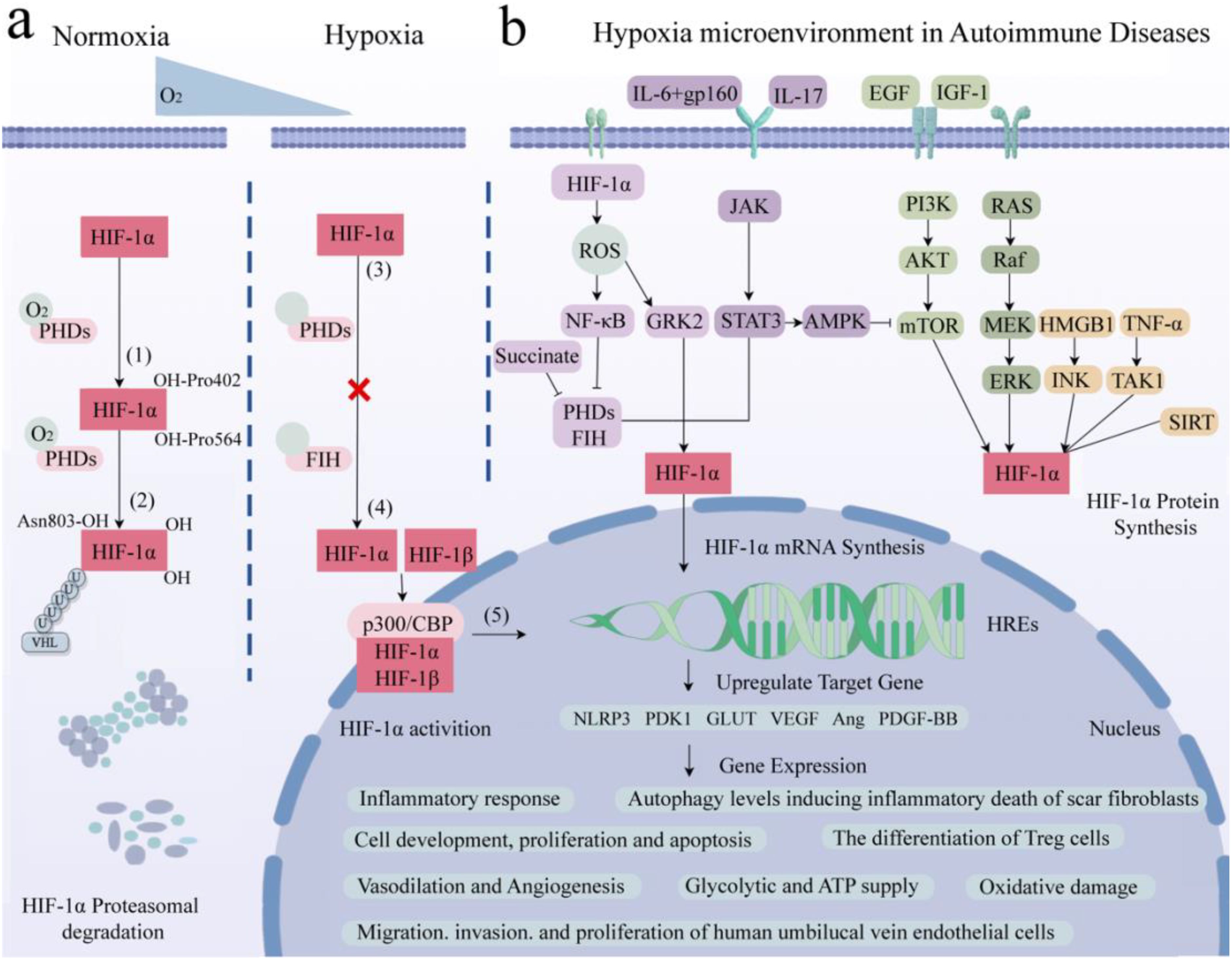
Figure 2. Role of HIF-1α in hypoxia-induced pathways and gene expression in AIDs. (A) Core molecule HIF-1α is proteasomal degradation (1). Two residues of HIF-1α, Pro402 and Pro564, were hydroxylated by PHDs under normal oxygen (2). The affinity between HIF-α and pVHL after hydroxylation is enhanced to form a stable complex. Attracts the accumulation of multiple Ub, resulting in HIF-1α degradation. (3) Hypoxia up-regulates intracellular ROS, inhibits PHDs and FIH, and HIF-1α accumulates in cytoplasm without degradation. (4) HIF-1α enters the nucleus and binds to HIF-1β and cofactor p300/CBP to form a stable heterodimer. (5) The dimer recognizes and binds to HRE and activates transcription of downstream genes. (B) Related signaling pathways and target gene expression in AIDs. AID patients have local hypoxic microenvironments, which regulate HIF-1α through the interaction of PHDs/HIF-1α/pVHL, NF-κB, JAK/STAT, PI3K/AKT/mTOR and MAPK signaling pathways. It is involved in inflammatory response, glucose metabolism and ATP supply, vasodilation and angiogenesis, inducing apoptosis, regulating autophagy level, promoting cell development and antioxidant damage. Created with figdraw.com.
2.3 Activation of HIF in hypoxic microenvironmentStudies have found that the classical mitogen-activation protein kinase (MAPK), including its regulated extracellular protein kinases (ERK) subfamily, play crucial roles in the hypoxic microenvironment, activating HIF-1α, protecting cell development, and avoiding oxidative damage (37). Additionally, epidermal growth factor (EGF) and insulin-like growth factor-1 (IGF-1) activate the phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB/AKT) signaling pathway, activating the mechanistic target of rapamycin kinase (mTOR). This cascade promotes HIF-1α expression and subsequently upregulates VEGF expression, ultimately promoting vasodilation (36). Furthermore, signal transducer and activator of transcription 3 (STAT3) can inhibit the mTOR/HIF-1α pathway by activating AMP-activated protein kinase (AMPK), leading to reduced cell proliferation and increased apoptosis (38). Meanwhile, Janus tyrosine kinase (JAK) can phosphorylate and activate downstream STAT3, upregulating nucleotide-binding oligomerization structure-like receptor family pyrin domain protein 3 (NLRP3) expression and thereby exacerbating inflammatory responses (39). In parallel, IL6 activates the STAT3/HIF-1α pathway through interaction with the gp160 protein, regulating the differentiation of Foxp3+ regulatory T cells (Treg). Conversely, IL-17 regulates autophagy levels through the STAT3/HIF-1α signaling pathway, ultimately inducing inflammatory death of scar fibroblasts (40–42).
It has been reported that tumor necrosis factor α (TNF-α) can bind with transforming growth factor-activated kinase 1 (TAK1), upregulating HIF-1α expression, thereby promoting glycolysis (43). Similarly, Hypoxia-induced high mobility group proteins 1 (HMGB1) can activate the c-Jun N-terminal kinase (JNK), which, in turn, stimulates the HIF-1α/VEGF signaling axis, facilitating angiogenesis (44). Additionally, the silence information regulator (SIRT) is a class of nicotinamide adenine dinucleotide+-dependent deacetylases. SIRT1 protects HIF-1α from deacetylation by binding to its inhibitory domain, while inhibiting SIRT2 downregulates fibroblast phosphorylation (45, 46). Overexpression of SIRT6 inhibits the ubiquitin proteasome system and promotes HIF-1α accumulation, upregulating the expression of VEGF, angiopoietins1 (Ang1), Ang2, and platelet-derived growth factor-BB (PDGF-BB), consequently enhancing migration, invasion, and proliferation of human umbilical vein endothelial cells (47, 48). This suggests that within the hypoxic microenvironment, the central regulatory factor HIF-1α is orchestrated by numerous signaling pathways apart from the oxygen sensor pathway PHDs/HIF-1α/pVHL, encompassing MAPK, PI3K/AKT/mTOR, and JAK/STAT among others (Figure 2B). Through intricate crosstalk with these diverse signaling cascades, HIF-1α modulates the secretion of inflammatory factors and participates in the regulation of various cellular processes, including survival, proliferation, metabolism, angiogenesis, and inflammatory responses.
Furthermore, in addition to the canonical activation of HIF-1α under hypoxic conditions through the aforementioned signaling pathways, this transcriptional factor can also be elicited by diverse non-hypoxic stimuli, including the accumulation of vitamin B1 (thiamine) deficiency (49), metabolic intermediates (notably succinate) (44), oxidative stress (32, 33), inflammatory responses, and metabolic reprogramming (50–52). Thiamine deficiency leads to an accumulation of metabolic intermediates, such as pyruvate and lactate, which stabilize HIF-1α through inhibition of prolyl hydroxylases (PHDs), thereby inducing HIF-1α-mediated gene expression (49). Succinate, serving as an upstream activator of HIF-1α, is translocated from the mitochondria to the cytoplasm, where it inhibits PHD activity, leading to the activation of HIF-1α. This, in turn, upregulates interleukin-1β (IL-1β) expression and triggers inflammatory responses (44, 53). Additionally, due to oxidative stress, intracellular reactive oxygen species (ROS) levels are elevated. ROS stimulate nuclear factor kappa-B (NF-κB), which downregulates PHDs and HIF, leading to cytoplasmic accumulation of HIF-1α and subsequent activation of downstream signaling molecules (32, 33). Increased ROS can induce upregulation of G protein-coupled receptor kinase 2 (GRK2)/HIF-1α and induce pyroptosis through the NLRP3 inflammasome-mediated pathway (54). Conversely, low levels of HIF-1α downregulate downstream target genes such as phosphoinositide-dependent protein kinase-1 (PDK1) and GLUT1, thereby regulating the glycolytic pathway and intracellular adenosine triphosphate (ATP) supply (35).
The Warburg effect, also known as aerobic glycolysis, denotes a significant intensification of the glycolytic pathway under aerobic conditions, characterized by augmented glucose uptake and lactate production (50–52). Under normal aerobic conditions, cells primarily depend on mitochondrial oxidative phosphorylation for energy production, with the glycolytic pathway exhibiting a relatively subordinate role. However, in the context of the Warburg effect, cells exhibit a notable enhancement of the glycolytic pathway even when oxygen is present, resulting in increased glucose consumption and lactate generation. This metabolic reprogramming is crucial for rapid cell proliferation and adaptation to hypoxic environments, frequently observed in tumors cell and inflammatory conditions. Lactate accumulation, a byproduct of enhanced glycolysis, has been shown to play a key role in immune regulation, particularly in pro-inflammatory responses. Lactate not only facilitates the metabolic reprogramming in various immune cells but also induces senescence and inflammation, promoting autoimmune pathological processes (55). The persistence of an autoimmune response can also be associated with the Warburg effect (50, 56). HIF-1α serves as a downstream effector of the mTOR signaling pathway, regulates the synthesis of diverse proteins and enzymes, modulates cellular responses to hypoxic conditions, and facilitates glycolytic metabolism (57, 58). It plays an important role in modulating the Warburg effect through the upregulation of glycolytic gene expression, particularly GLUT (35), hexokinase, and lactate dehydrogenase (LDH), which enhances cellular glucose uptake, utilization, and lactate production. Research have shown that PDGF activates the PI3K/AKT/mTOR/HIF-1α signaling pathway, thereby enhancing the Warburg effect in pulmonary artery smooth muscle cells (59). MARK kinases, members of the AMPK-related kinase family, potentiate the AMPKα1/mTOR/HIF-1α signaling axis, contributing to the Warburg effect and cell proliferation in non-small cell lung cancer (60). We hypothesize that in the hypoxic microenvironment, the activation of HIF-1α, via the mTOR signaling pathway, may further exacerbate the Warburg effect, stimulating cell proliferation and adaptation. Additionally, this augmentation may potentially exacerbate autoimmune pathological processes associated with hypoxia.
Collectively, the complex interactions between HIF-1α and its diverse hypoxia-dependent and non-hypoxia-stimulated regulatory pathways form a specific, multifaceted hypoxia-responsive pathway that is critical for cellular adaptation to changing environments and for orchestrating inflammatory responses. This understanding of HIF-1α’s versatility and complexity provides valuable insights into the development of therapeutic strategies targeting this pathway for the treatment of inflammation-related diseases.
3 Hypoxic microenvironment and immune cells3.1 NeutrophilsNeutrophils are the core cells of innate immunity. Their overactivation results in accumulation in inflammatory areas, where they generate ROS, activate proteases of soluble proteins, induce innate and adaptive immune responses, release neutrophil extracellular traps (NETs), mediate gene expression and cell signaling, cellular metabolism, ultimately precipitating local tissue damage (61). It has been reported that neutrophils in the peripheral blood and synovial fluid of RA patients can promote ROS generation. Inflammatory cytokines secreted in the inflammatory area, such as granulocyte-macrophage colony stimulating factor (GM-CSF), TNF-α, IL-1β, and interferon, can activate HIF-1α to inhibit neutrophil apoptosis (62, 63). Hypoxia can promote the activation of NF-κB and HIF-1α in the joints of RA patients, inhibit cell apoptosis, regulate neutrophil cytoplasmic retention, and thus prolong RA inflammation (64). Additionally, research has demonstrated abnormal ROS generation in neutrophils in the peripheral blood of SLE patients, with elevated levels of O2, H2O2, and HO compared to normal individuals (65). This indicates the involvement of HIF-1α in neutrophil aggregation and secretion of inflammatory factors. Belimumab, approved in the UK for the treatment of SLE patients, inhibits B-lymphocyte stimulator (BLyS) primarily stored in neutrophils (66). In RA patients, disease-modifying anti-rheumatic drugs (DMARDs) targeting TNF significantly downregulate the expression of neutrophils and activate NF-κB (67). JAK inhibitors, such as Baricitinib and Tofacitinib, are small molecule drugs that hinder neutrophil migration and ROS generation by suppressing the secretion of inflammatory factors (68). This implies that hypoxic microenvironment can enhance the pro-inflammatory activity of neutrophils, increase oxidative stress and tissue damage, which may play a role in the inflammatory response of multiple AIDs. Modulating the local hypoxic microenvironment in AIDs to curb neutrophil activation and attenuate target organ damage is also one of the mechanisms by which biological agents exert their effects.
3.2 MacrophagesIn a hypoxic environment, the activation of HIF-1α specifically promotes glycolysis in monocytes and macrophages, thereby enhancing their antigen presentation capabilities and secretion of inflammatory factors (69). Furthermore, the upregulation of HIF-1α in macrophages contributes to the clearance of infections by enhancing bactericidal effects against pathogens such as mycobacterium tuberculosis (70). Macrophages are categorized into pro-inflammatory M1-type and anti-inflammatory M2-type, both of which participate in the activation of HIF-1α under certain conditions (71, 72). Research has demonstrated that HIF-1α can promote the expression of NF-κB in macrophages induced by lipopolysaccharide (LPS) (73). Following the activation of pro-inflammatory M1 macrophages, the activation of NF-κB, upregulation of HIF-1α, and the presence of ROS synergistically contribute to the polarization of macrophages towards the M1 phenotype (74). Blocking the NF-κB signaling pathway in HIF-1α promotes the transition of macrophages from M1 to M2 phenotype, which is crucial for the treatment of RA (75). In addition, Jing revealed that immunoglobulin G (IgG) stimulation of human and murine macrophages results in the production of numerous proinflammatory mediators, including IL-1β, which is dependent on HIF-1α and mTOR signaling, leading to metabolic reprogramming and subsequently driving inflammation in AIDs, such as RA, SLE, SSs, Sjögren’s syndrome (SS) and vasculitis (76). Collectively, these findings suggest that inhibiting metabolic reprogramming of macrophages and downregulating HIF-1α expression in the hypoxic microenvironment may represent viable therapeutic strategies for alleviating tissue inflammation in patients with AIDs.
3.3 Dendritic cellsResearch has found that in the hypoxic microenvironment, HIF-1α can directly bind to long non-coding ribonucleic acid Dpf3 (LncRNA Dpf3) or p38 MAPK, subsequently inhibiting the glycolytic metabolism and migratory potential of dendritic cells (77). Concurrently, the interaction of HIF-1α with the PI3K/AKT pathway enhances the migratory abilities of dendritic cells (78), revealing its dual roles in different signaling pathways. Additionally, downregulation of HIF-1α can inhibit its antifungal immune response (79), further emphasizing its critical role in immune defense. Some findings reveal that the absence of HIF-1α impairs the ability of dendritic cells to induce regulatory Treg, resulting in exacerbated intestinal inflammation in mice (80). Additionally, the downregulation of HIF-1α markedly diminishes the level of granzyme B released by DCs during T cell activation (81), highlighting the indispensable role of HIF-1α in facilitating DC-mediated T cell activation. Other studies have also demonstrated that HIF-1α exerts a suppressive effect on IL-12 production in dendritic cells, thereby restricting the development of Th1 cells and subsequently influencing the balance of immune responses (82).Collectively, these findings suggest that the hypoxic microenvironment, by modulating the expression of HIF-1α, may regulate dendritic cell migration capability, glucose metabolism, immune defense capabilities, and T cell activation, thereby exerting profound effects on disease progression.
3.4 Natural killer cellsFurthermore, research has revealed that under hypoxic conditions, IL-15 activates the STAT3 signaling pathway, while IL-2 stimulates the PI3K/mTOR signaling pathway, thereby maintaining HIF-1α stability and enhancing the innate immune defenses of natural killer cells against microbial infections and cancer (83, 84). Moreover, during cytomegalovirus infection, downregulation of HIF-1α expression has been shown to impact the survival of natural killer cells (85). This indicates that regulating natural killer cells within the hypoxic microenvironment may be involved in anti-tumor and antiviral processes.
3.5 B cellsIn the hypoxic microenvironment, HIF-1α can regulate B cell development, differentiation, maturation, and antibody secretion (86). It has been shown that oxygen level plays a critical role in regulating B cell differentiation, where hypoxia promotes differentiation into unique CD27++ B cells with enhanced antibody secretion capacity upon restimulation (87).HIF-1α is a key regulator of B cell glycolytic metabolism in the germinal center, and low expression of HIF-1α inhibits B cell responses, leading to abnormal class switching (88). Additionally, HIF-1α serves as a critical transcription factor for B cells producing IL10 in AIDs. Downregulation of HIF-1α significantly reduces the number of IL-10 secreting B cells, exacerbating collagen-induced arthritis and experimental autoimmune encephalomyelitis (EAE) (89). It has been speculated that the use of HIF activators in clinical practice may improve the local hypoxic microenvironment and regulate B cell-related AIDs.
3.6 T cellsUnder hypoxic conditions, the T cell receptor (TCR) engages with HIF-1α, thereby activating HIF-responsive genes. This interaction modulates the equilibrium among Th1, Th2, Th17, Treg, and other T cell subsets, subsequently regulating inflammatory responses (90–92). Investigations have revealed that, under hypoxic conditions, in antigen-presenting cells lacking HIF-1α, overexpression of STAT3 significantly inhibits Th1 proliferation (93). Conversely, micro-ribonucleic acid (microRNA) 182, an inhibitor of HIF-1α, expedites Th1 cell differentiation, potentially becoming a novel therapeutic target in the progression of autoimmune inflammation (94). During pathogen infection, HIF-1α expression is upregulated in Th2 cells, thereby fostering their proliferation (95). Downregulation of HIF-1α inhibits Th0 differentiation into Th17 cells and reduces the expression of the Th17 transcription factor RAR-related orphan receptor gamma t (RORγt) (96). HIF-1α regulates the development of Treg and Th17 cells by directly transcribing and activating RORγt expression and inhibiting FoxP3 transcriptional activity (97). Furthermore, research has found that HIF-1α promotes the development of Th17 cells while inhibiting the differentiation of Treg cells (98). HIF-1α upregulates the expression of CD73 in Treg cells, enhancing their suppressive capabilities through CD73 binding and facilitating a metabolic shift towards aerobic glycolysis (99). Elevation of O2 levels at tumor sites downregulates HIF-1α, affecting the metabolism of tumor cells and Treg cells, and ultimately reducing cancer cell proliferation (100). Additionally, in SLE patients’ T cells, SIRT2 promotes Th17 cell differentiation and inhibits CD4+ T cell production of IL-2, correcting abnormal expression of IL-17A and IL-2, and benefiting CD4+ T cells in lupus prone mice and SLE patients (101). It is speculated that targeting HIF-1α to regulate the differentiation, metabolism, and function of T cells in a low oxygen microenvironment may have potential therapeutic value for immune responses, inflammation, tumors, and AIDs (Figure 3).
Figure 3. Activation of the core molecule HIF and immune cells in hypoxic microenvironment. The core molecule HIF in hypoxic microenvironment affects neutrophils, macrophages, dendritic cells, natural killer cells, B cells and T cells, mediates congenital and adaptive immune responses, and participates in cell development, differentiation and maturation, gene expression, metabolic reprogramming, anti-tumor, prevention of viral infection, and AIDs. Created with figdraw.com.
3.7 Myeloid-derived suppressor cellsMDSCs, initially identified within the tumor microenvironment, represent a heterogeneous population of immature myeloid cells, comprising diverse stages of myeloid differentiation, such as myeloid progenitor cells, immature macrophages, monocytes, and dendritic cells. MDSCs exert their immunosuppressive effects by inhibiting the function of T cells, B cells, natural killer cells, and other myeloid lineages, thereby dampening immune responses and facilitating tumor progression through the suppression of antitumor immune responses (102, 103). Under normal physiological conditions, myeloid progenitor cells undergo ordered differentiation into mature granulocytes, macrophages, and dendritic cells, which are then distributed throughout various tissues and organs to execute and maintain normal immune functions. However, under pathological conditions such as tumor development, bone marrow transplantation, severe trauma, infection, inflammatory immune responses, and various AIDs, the expression of immunosuppressive factors is significantly upregulated, inhibiting the normal differentiation of myeloid progenitor cells and leading to abnormal expansion and accumulation of MDSCs. This, in turn, disrupts the balance of the immune system and exacerbates the persistence and progression of pathological states (104–108).
Recent studies have elucidated that the immunosuppressive function of MDSCs is intricately regulated by a myriad of complex metabolic pathways, with hypoxia playing a pivotal role in this regulation (109–111). HIF-1α orchestrates metabolic reprogramming in MDSCs, particularly by augmenting their glycolytic capacity, a phenomenon known as the Warburg effect. This metabolic shift enables MDSCs to sustainably generate energy in hypoxic environments, while concurrently mitigating ROS production and subsequent apoptosis, thereby safeguarding their survival and functionality (110, 112).Chiu et al. have revealed that in MDSCs derived from hepatocellular carcinoma patients, hypoxia activates HIF-1, leading to the upregulation of ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2) expression, which subsequently facilitates the enzymatic hydrolysis of extracellular ATP into 5’-AMP. This metabolic alteration, in turn, impedes the normal differentiation of MDSCs and fosters their accumulation within the tumor microenvironment (113). This underscores the significance of MDSCs as a crucial immunosuppressive cell population, whose accumulation and activation within the tumor microenvironment constitute a vital component of tumor immune evasion mechanisms. We hypothesize that inhibiting the activity of ENTPD2 under hypoxic conditions may effectively disrupt MDSC function, thereby restoring T-cell activity and function, with the potential to enhance the overall efficacy of immunotherapy.
Furthermore, HIF-1α exerts profound effects on the recruitment, activation, and immunosuppressive functions of MDSCs. Li et al. have demonstrated that TGF-β enhances the immunosuppressive capabilities of MDSCs in non-small cell lung cancer patients by activating the mTOR/HIF-1 signaling pathway, which in turn upregulates the expression of CD39 and CD73 on MDSCs (114).We postulate that the development of small molecule inhibitors or antibodies specifically targeting HIF-1α activity, or the utilization of existing mTOR inhibitors (such as rapamycin and its derivatives), may effectively impede the activation of HIF-1α by TGF-β, thereby inhibiting the recruitment and activation of MDSCs. This strategy offers novel insights and potential therapeutic avenues for alleviating immunosuppression and controlling tumor progression. Ding et al. have devised hypoxia-responsive sono-activatable semiconducting polymer nanopartners (SPNTi), which are capable of generating singlet oxygen (1O2) during sonodynamic therapy (SDT), thereby disrupting the surface shell to release hypoxia-responsive drugs (tirapazamine (TPZ) conjugates) and MDSC-targeting drug (ibrutinib) (115). In the severely hypoxic tumor microenvironment, the TPZ conjugates are activated to augment immunogenic cell death (ICD), while ibrutinib effectively mitigates the immunosuppressive effects of MDSCs. The synergistic effects of these two agents significantly potentiate antitumor immune responses (115). This innovative strategy overcomes the limitations posed by the hypoxic environment post-treatment, enabling precise targeting of MDSCs and presenting a novel, high-precision, and safe sonodynamic immunotherapy approach for cancer treatment.
Studies have demonstrated that MDSCs accumulate prominently in secondary lymphoid organs of AID patients, particularly at sites of disease manifestations such as RA, MS, and SLE (116–118). This revelation underscores the pivotal role of MDSCs in AID progression and provides a novel perspective for considering them as potential therapeutic targets. In both AID patients and corresponding animal models, the expansion and accumulation of MDSCs protect host tissues from damage caused by excessive T cell responses by inhibiting T cell activation and immune reactions (102, 119), This further indicates that MDSCs may exert a detrimental impact on immune responses by modulating the functions of CD4+ and CD8+ T cells, thereby contributing to the pathological processes of AIDs. Consequently, the development of targeted therapies aimed at precisely inhibiting the immunosuppressive functions of MDSCs at specific time points and tissue locations holds promise as a novel and critical strategy for restoring effective immune responses and promoting disease remission (118–121). In summary, hypoxia plays a pivotal role in the metabolic reprogramming, recruitment, activation, and immunosuppressive functions of MDSCs through HIF-1α. We speculate that inhibiting HIF-1α or key enzymes in the glycolytic pathway may attenuate the immunosuppressive functions of MDSCs, thereby altering the balance of immune responses and potentially contributing to the immune evasion mechanisms of AIDs.
4 Hypoxic microenvironment in autoimmune diseases4.1 Rheumatoid arthritisRA is a chronic and systemic AID, exhibiting a global prevalence ranging from 0.5% to 1.0%. The prevalence rate in mainland China is 0.42%, and it is hypothesized that about 5 million people are affected, with a male-to-female ratio of about 1:4. Its main pathological features encompass abnormal synovial inflammation proliferation, angiogenesis, pannus formation, as well as irreversible damage to cartilage and bones, ultimately resulting in joint deformity and disability (101, 122, 123). The pathogenesis of RA remains exceedingly intricate and is yet to be fully unraveled. In recent years, the importance of hypoxic microenvironment in the pathogenesis of RA has gradually attracted attention. The hypoxic microenvironment is the initiating factor for abnormal synovial proliferation, and it is involved in the important processes of RA occurrence and development (122–125). The synovium of RA patients frequently exhibits a hypoxic state, primarily attributable to the excessive proliferation of synovial tissue coupled with inflammatory reactions. The formation of new blood vessels is often insufficient to provide adequate oxygen to the synovium, leading to a decrease in local microenvironmental oxygen tension and the formation of a hypoxic synovial microenvironment, which in turn affects angiogenesis, inflammatory responses, matrix degradation, cartilage erosion, energy metabolism disorders, and oxidative stress (126, 127). This underscores the notion that local tissue hypoxia can exacerbate inflammation and immune dysfunction, further aggravating tissue hypoxia, thereby perpetuating a vicious cycle. Ahn et al. reported strong expression of HIF-1α in the intimal layer of macrophage-like synoviocytes (MLS) in human RA synovium (128), while HIF-2α is mainly expressed in fibroblast-like synoviocytes (FLS) in human RA synovium and collagen-induced arthritis model (CIA) (129). Related research found that compared to osteoarthritis (OA) patients, the expression of HIF in FLS and MLS of RA patients increased (128, 130). Moreover, HIF expression has been observed in chondrocytes and osteoclasts, indicating its ubiquitous presence in synovial tissues (129). Collectively, these findings suggest that distinct HIF subtypes are expressed in various synovial cell types, all of which participate in the occurrence and progression of RA.
Research has reported that the activation of PI3K/AKT/HIF-1α or NF-κB/HIF-1α pathways can stimulate the migration and invasion of FLS in RA patients (131, 132). The STAT3/HIF-1α/fascin-1 axis plays an important role in the hypoxic microenvironment of RA synovium, facilitating endothelial-to-mesenchymal transition (EndoMT) of FLS and thereby promoting migration and invasion (133). Under hypoxic microenvironment, HIF-1α regulates the migration and invasion of RA FLS and angiogenesis through the Notch-1 pathway (134). Furthermore, chemokine (C-X-C motif) ligand 12 (CXCL12) functions as a potent chemotactic and angiogenic factor, playing a role in angiogenesis. Hypoxia at the site of inflammation stimulates FLS to produce messenger RNAs such as CXCL12 and VEGF, resulting in a 50% and 132% increase in the protein levels of VEGF and CXCL12, respectively, after 24 hours of hypoxia (135). VEGF, the primary angiogenic target of HIF-1α, exhibits oxygen-dependent expression. The downregulation of HIF-1α notably diminishes VEGF-mediated angiogenesis in FLS. The activation of the PI3K/AKT/mTOR signaling pathway in the adjuvant arthritis model may be involved in inducing newly formed synovial vessels (136). Collectively, these findings suggest that HIF-1α is intricately involved in the proliferation, migration, and invasion of FLS in RA patients, alongside regulating angiogenesis and inflammatory responses.
As the disease progresses, joint erosion and bone degradation gradually intensify, representing another major characteristic of RA. The elevated expression of HIF-1α has been correlated with augmented bone erosion in RA patients (137). Knowles et al. have demonstrated that HIF-1α promotes bone resorption mediated by osteoclasts (OCs), while angiopoietin-like 4 (ANGPTL4) compensates for HIF-1α’s inadequate stimulation of OC activity and promotes the proliferation and differentiation of osteoblasts (OBs) (138). Swales et al. demonstrated that ANGPTL4 is highly expressed in OCs of RA patients in a HIF-1α-dependent fashion, contributing to bone resorption. In RA, HIF-1α induces the upregulation of receptor activator of NF-κB ligand (RANKL) in OBs, ultimately facilitating OCs-mediated bone resorption (137, 139). HIF-1α regulates the expression of Notch-1, thereby stimulating the production of matrix metalloproteinase 2 (MMP2) and MMP9 in endothelial cells (132). The interaction between HIF-1α and NF-κB promotes the enzymatic activity of MMP2 and MMP9, leading to the disruption of tissue barriers and bone materials (130). IL-38 has the potential to suppress inflammation and angiogenesis in CIA rats, upregulating osteogenic factors through the SIRT1/HIF-1α signaling pathway, thus mitigating joint damage (140). Collectively, these findings underscore the important role of HIF-1α in mediating bone destruction in FLS of RA patients.
Li et al. reported that the combination of dexamethasone (DEX) and artesunate (ART) can be used to treat RA by regulating the HIF-1α/NF-κB signaling pathway, controlling ROS clearance, and reversing macrophage polarization (75). Meanwhile, previous research findings indicate that berberine inhibits the proliferation and adhesion of FLS in RA patients, accomplishing this through modulation of the MAPK/FOXO/HIF-1α signaling pathway (141). Furthermore, Jia et al. discovered that administering roburic acid (RBA), a component of the anti-RA herb Gentiana, to RA joints can effectively block the ERK/HIF-1α/GLUT1 pathway, fostering the transition from M1 to M2 macrophage phenotype, suppressing inflammatory cytokines, and enhancing tissue repair, thereby yielding potent therapeutic outcomes for RA (142). Furthermore, Pang et al. recently found that the expression of receptor-interacting protein kinase 3 (RIPK3) is significantly increased in intestinal epithelial cells (IECs) of RA patients. Their findings reveal that HIF-1α can induce a switch from physiological cell apoptosis to pathological necrotic apoptosis by transcriptional inhibition of RIPK3, thereby initiating intestinal mucosal inflammation and exacerbating arthritis. Utilizing HIF-1α inhibitors, such as Roxadustat, has been shown to block IEC death, maintain intestinal barrier function, and ameliorate arthritic symptoms (143, 144). This further indicates that modulating HIF activity or inhibiting the inflammatory response of FLS in RA patients under hypoxic conditions may provide new insights for RA patient treatment strategies. This underscores the potential for modulating HIF activity or inhibiting the inflammatory response of FLS in RA patients under hypoxic conditions, which may potentially yield novel therapeutic avenues for RA treatment strategies.
Increasing evidence suggests that cells derived from the mesenchyme, especially FLS, are crucial cellular elements in RA and promising targets for future arthritis treatments. They play a critical role in mediating direct tissue damage and the continuation of complex disease processes in autoimmune joint diseases like RA. It is speculated that targeting the regulation of HIF-1α expression potentially modulate the behavior of FLS within local hypoxic microenvironments, thereby exerting a positive influence on inflammation, angiogenesis, and bone destruction in RA patients. Consequently, strategies aimed at modulating HIF-1α to ameliorate local hypoxic conditions may hold significant therapeutic potential for patients with AIDs (Figure 4).
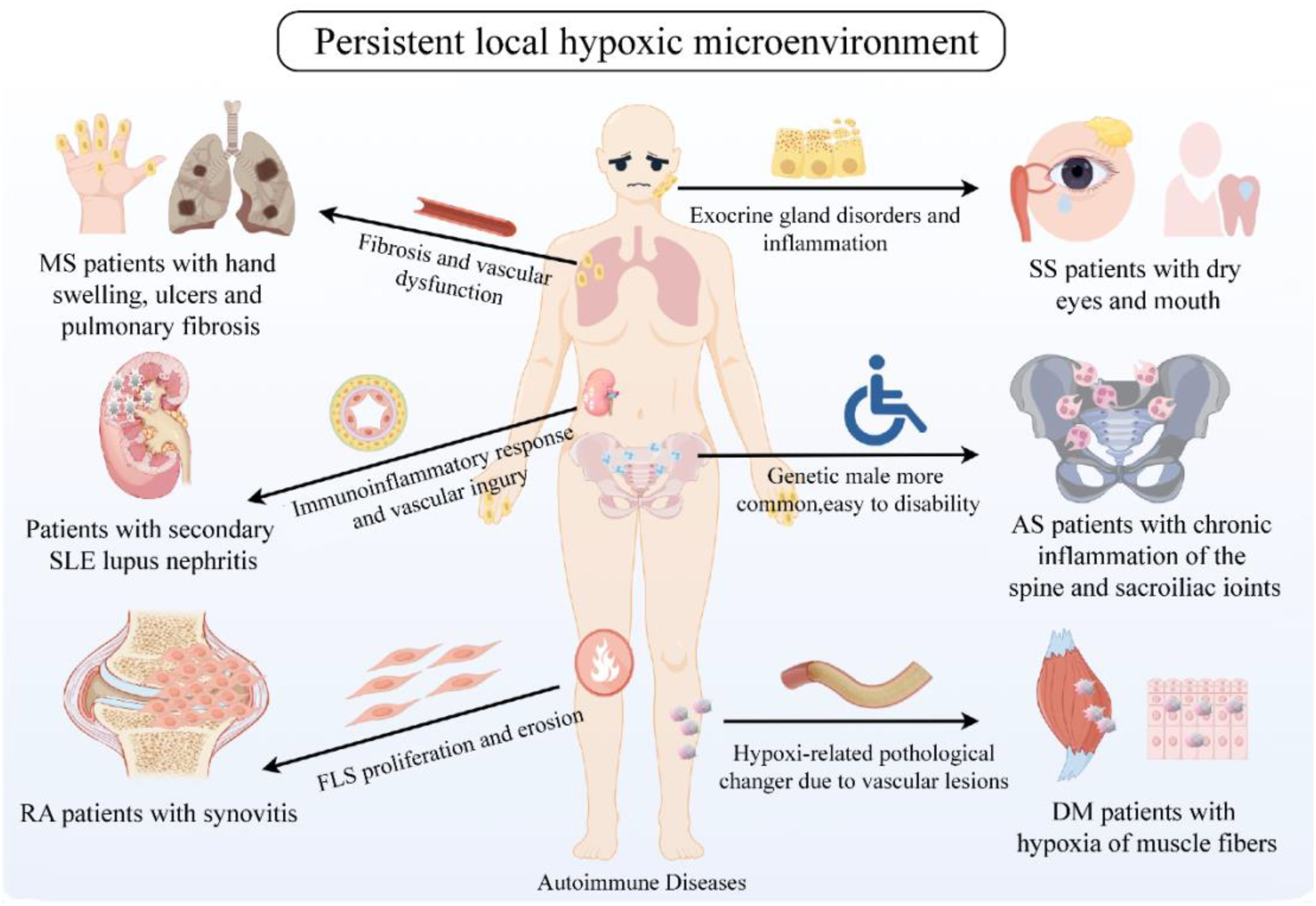
Figure 4. Persistent local hypoxic microenvironment in clinically common AID patients. Clinically common AIDs include RA, SLE, MS, DM, SS, AS, etc. The common feature is the continuous hypoxic microenvironment of patients. Hypoxia affects the proliferation and erosion of tissue cells, inflammatory response, vascular damage, glandular dysfunction, skin and bone fibers, etc., thus participating in the pathogenesis of AIDs. Created with figdraw.com.
4.2 Systemic lupus erythematosusSLE, a prototypical AID, is characterized by recurrent autoantibody production, complement system activation, and immune complex deposition across multiple organ systems, notably lupus nephritis (LN) (145–147). Recent research has increasingly illuminated a close association between the hypoxic state in SLE patients and its occurrence and development (148). From a pathological perspective, SLE often exhibits a hypoxic state in local tissues due to abnormal activation of T and B cells, sustained (148). Moreover, metabolic reprogramming in immune cells, particularly mitochondrial dysfunction, has been linked to enhanced immune responses and tissue damage in SLE, further highlighting the role of hypoxia in driving autoimmune pathology (149). Pathologically, SLE frequently exhibits a hypoxic state in local tissues due to abnormal activation of T and B cells, sustained immune-inflammatory reactions, and vascular damage. This hypoxic state modulates SLE pathogenesis of SLE through mechanisms that include altering immune cell metabolism, enhancing inflammatory mediator production, and regulating angiogenic factor expression. Caielli et al. observed that HIF degradation defects during erythrocyte maturation in SLE patients result in the accumulation of mitochondria-dense red blood cells and subsequent mitochondrial dysfunction, contributing significantly to SLE pathogenesis (150). Another investigation revealed that in photosensitive SLE patients, circulating miR-210 and HIF-1α levels are significantly elevated and positively correlated (r=0.886, P<0.05), and miR-210, a critical hypoxia-related molecule, can regulate hypoxic signaling within hypoxic microenvironments (151, 152). Collectively, these findings underscore a close association between the hypoxic microenvironment and the initiation, progression, and severity of SLE.
Recent studies have revealed that mitochondrial activity not only influences cellular energy metabolism but also affects epigenetic modifications, such as histone methylation and acetylation, which are crucial in the regulation of immune responses under hypoxic conditions. This interaction may contribute to long-range effects on autoimmune disease progression by altering gene expression and immune cell function (153). Research has demonstrated that CD4+ T cell proliferation is apparent in both the peripheral blood of SLE patients and the renal tubulointerstitial compartment of individuals with proliferative lupus nephritis (PLN). These cells are capable of producing high levels of mitochondrial ROS (mtROS) in a process known as reverse electron transfer (RET). A key feature of the RET process is the accumulation of succinic acid. CD4+ T cells produce and secrete higher levels of succinic acid, upregulating HIF-1α, which further affects their metabolism and function, including promoting inflammation and immune response (44, 75, 101, 154). This suggests that CD4+ T cells play an important role in AIDs such as SLE through succinic acid accumulation, HIF-1α activation and mtROS production. We speculate that interventions targeting the activation pathway of CD4+ T cell subpopulations, such as down-regulation of HIF-1α or inhibition of hypoxic microenvironment, may reduce the expansion and function of CD4+ T cells, thereby inhibiting the initiation or persistence of SLE. In T cells of SLE patients, mTOR activation triggers ROS production, while SIRT2 concurrently upregulates IL-17A expression in these cells by modulating the mTOR complex 1 (mTORC1)/HIF-1α/RORγt signaling pathway, thereby contributing to the pathogenesis of SLE (101, 155). We speculate that downregulation of HIF-1α via the mTOR1 pathway or alleviation of the hypoxic microenvironment may attenuate the Warburg effect, impede T cell differentiation, inhibit glycolytic metabolism, and modulate inflammatory and immune responses in AID patients such as SLE.
In the hypoxic microenvironment, AKT and mTOR are located upstream of HIF-1α (36). Kshirsagar et al. discovered that downregulation of mTOR in LN patients leads to a reduction in STAT3 and Th17 cells within effector T cells, while inhibiting the AKT signaling pathway hinders the migration of Th17 cells towards inflammatory sites, effectively mitigating the chronic inflammatory process in LN (38, 156). The activation of mTORC1 precedes the onset of SLE and its associated complications, including antiphospholipid syndrome, potentially serving as an early diagnostic biomarker for the disease. The targeting of mTORC1 with mTOR inhibitors to counteract its overactivation may replace drugs with significant side effects such as prednisone or cyclophosphamide (151). It has been reported that in renal tissue macrophages, the induction of glycolysis through the mTOR/HIF-1α signaling pathway exerts an inhibitory effect on inflammation and the recruitment of neutrophils mediated by immune complexes, underscoring its important role in diseases such as LN (76). Additionally, luteolin has been shown to inhibit the expression of HIF-1α and oxidative stress in macrophages, thereby mitigating kidney damage attributed to macrophage infiltration in LN (157). Chen et al. pointed out that renal hypoxia contributes to heightened T cell infiltration and kidney damage, partially mediated by HIF-1α-induced metabolic reprogramming in lupus-prone mice (MRL/lpr mice). Targeting HIF-1α may represent a viable therapeutic approach for mitigating kidney damage in autoimmune disorders, including AIDs (158). Collectively, these findings suggest that modulating key factors within the HIF-1α signaling pathway, both upstream and downstream, can effectively optimize effector cell metabolism, diminish the proliferation and overactivation of abnormal, self-reactive cells, and subsequently ameliorate tissue inflammation in SLE patients, particularly those with LN.
Current therapeutic approaches for SLE primarily focus on the suppression of the overall immune response, utilizing medications such as corticosteroids, hydroxychloroquine, and azathioprine (158). Nonetheless, the prolonged use of corticosteroids and other forms of immunosuppression frequently results in significant adverse effects, notably infections, which have emerged as a leading cause of mortality in SLE patients (159). Targeting tissue-infiltrating immune cells has been shown to be efficacious in mitigating organ damage (160). Research has demonstrated that the utilization of HIF-1α inhibitors (PX-478) in LN mouse models effectively delays the recruitment of T cells to the kidneys, reverses the detrimental effects of T cells on tissue, and ameliorates kidney damage (158). We speculate that inhibiting HIF-1α to modulate the hypoxic microenvironment may offer novel insights into the development of therapeutic strategies for SLE.
4.3 Multiple sclerosisMS is a systemic AID of unknown etiology, characterized by immune dysregulation, skin and visceral organ fibrosis, and vascular dysfunction, which are thought to be related to the pathogenesis of MS (161–163). Hypoxia is an important factor in triggering fibrosis, inducing the deposition of extracellular matrix (ECM) and increasing VEGF expression. It promotes tissue fibrosis by binding to PDGF receptors, while excessive ECM deposition exacerbates vascular lesions and hypoxia, further aggravating fibrosis (162, 164, 165). Ottria et al. found that hypoxia can drive the production of CXCL4 in MS patients’ plasmacytoid dendritic cells by stabilizing HIF-2α through excessive production of mtROS (P=0.0079) (166). This finding underscores the potential therapeutic value of inhibiting the mtROS/HIF-2α signaling pathway and ameliorating the hypoxic microenvironment to attenuate the detrimental effects of CXCL4 in MS and other CXCL4-driven inflammatory disorders. Additionally, Liu et al. found that hypoxia promotes MS fibroblast activity through autophagy acceleration, while autophagy-mediated hypoxia-induced 2-methoxyestradiol inhibits MS collagen synthesis and EndoMT (167). Abnormal expression and activity of HIF-1α have also been implicated in the development of various fibrotic diseases under hypoxic microenvironment conditions, including renal and cardiac fibrosis (168). Specifically, HIF-1α in human coronary artery endothelial cells has been shown to induce EndoMT and promote cardiac fibrosis through snail activation (169). Similarly, HIF-1α-mediated EndoMT leads to pulmonary fibrosis (170). Li et al. further confirmed that hypoxia participates in the pathogenesis of MS by modulating EndoMT-related genes, potentially involving transforming growth factor-β (TGF-β), NF-κB, TNF, and mTOR signaling pathways (171). Collectively, these findings emphasize the role of the hypoxic microenvironment in MS-associated fibrosis, suggesting that its regulation may represent a potential new therapeutic strategy for MS and other fibrotic diseases.
At the early stage of MS pathogenesis, vascular endothelial damage and apoptosis upregulate vasoactive substances, prompting the migration of immune cells towards the vasculature and peripheral tissues, leading to inflammation, oxidative stress, and tissue hypoxia (162, 172, 173). In turn, this inflammatory milieu, characterized by the production of inflammatory and pro-fibrotic cytokines, further exacerbates microvascular endothelial cell injury and apoptosis (174, 175), creating a vicious cycle that culminates in thrombosis, inflammation, and a myriad of clinical manifestations ranging from skin to parenchymal organs (174, 176). These observations underscore the involvement of the hypoxic microenvironment in MS-associated vascular pathology, implying that strategies aimed at improving vascular oxygenation, mitigating immunoinflammatory damage, downregulating vasoactive substances, and reversing vascular abnormalities may reduce ischemic damage to the skin and visceral organs.
Moreover, approximately 20%-40% of MS patients clinically develop subepidermal calcium salt deposits, known as cutaneous calciphylaxis (161, 177, 178). Hypoxia has been posited as a possible link between vascular dysfunction and calciphylaxis in MS (179), where vascular dysfunction with defective angiogenesis leads to recurrent episodes of ischemia-reperfusion injury, increasing the products of oxidative stress, which results in tissue hypoxia (180). Hypoxia accompanied by damage to collagen, elastin or subcutaneous fat further exacerbates tissue necrosis, releasing denatured proteins and leading to calcification (181). Compared to healthy individuals, MS patients, both with and without calciphylaxis, exhibit elevated basal expression levels of hypoxia-related proteins (GLUT1 and VEGF), with the highest levels observed in those with calciphylaxis (182). Vascular variations may predispose MS patients to finger ischemia and hypoxia-induced limb osteolysis, thereby facilitating the deposition of calcium plaques in the skin tissues (178). These observations further suggests that the hypoxic microenvironment is closely associated with the development of cutaneous calciphylaxis in MS patients and is involved in their vascular pathology and calcium salt deposition.
Given the association between hypoxia and MS pathogenesis, it is tempting to speculate that regulating HIF-1α may be possible to improve the local hypoxic microenvironment in MS patients, reducing vascular inflammation and fibrosis, and ultimately alleviating clinical symptoms.
4.4 DermatomyositisDM, an inflammatory myopathy that primarily affects skin and muscle tissues, is prevalent among young individuals and adults. Changes related to hypoxia induced by vascular alterations may be a significant pathogenic mechanism (183, 184). Recent studies have demonstrated that in DM muscle fibers, endothelial cells prominently express HIF-1α and HIF-1β, while the muscle fibers themselves exhibit expression of HIF-2α, as well as VEGF, and VEGF receptors (185). VEGF is involved in hypoxia regeneration and angiogenesis, and the upregulation of hypoxia-related proteins may be
留言 (0)