
記住我


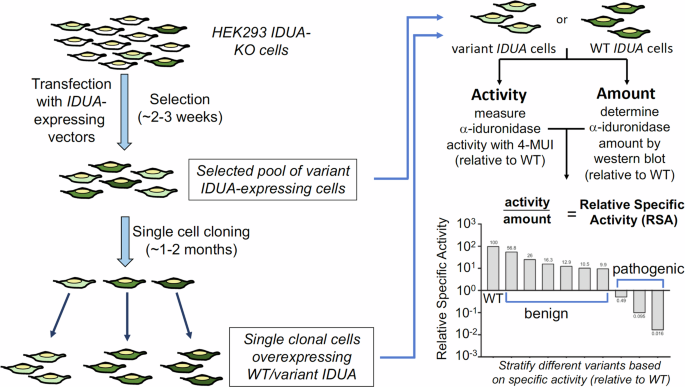
We focused on alternative splicing events that differentiate between a subset of a gene’s transcripts and the rest of its transcripts. We examined rates of exon inclusion/exclusion in comparison to the overall rate of gene expression in nine tissues with 226 to 653 samples each (Fig. 1, Supplementary Table 1).
Fig. 1: Multitissue RNA-seq analysis identifies association between gene expression and alternative splicing.
A RNA-seq samples from 9 tissues with the largest number of samples were analyzed. B For each of 141,043 alternative splicing events with above-threshold variability in the nine tissues, total gene expression and percent-spliced in (ψ) were calculated and logistic regression was performed to test the association of gene expression and ψ. The cartoon at the top shows the regions of the introns surrounding the cassette exon that were investigated bioinformatically. C 3667 UHP (for “upregulated-high ψ”) exons with a statistically significant positive association were identified (ψ increases as total gene expression increases). One example is shown, exon 2 of ABI2. D 3207 DHP (for “downregulated-high ψ”) exons with a statistically significant negative association were identified (ψ decreases as total gene expression increases). In the example, exon 4 of ABLIM2 is shown. E We hypothesized that our observations are related to mechanisms including coupling of RNAP2 extension speed with splicing decisions. In this example, a relatively fast RNAP2 elongation rate exposes a regulatory element (red box) at the 3′ end of intron B (shown in yellow), which promotes skipping of exon B (left); in contract, slower RNAP2 elongation fails to expose this element for a period of time sufficient for the splicing machinery to include exon B. This is one of many mechanisms that link transcription and alternative splicing. The figure was generated using ggpubr and Adobe Illustrator.
Type 0, upregulated-high ψ (UHP), and downregulated-high ψ (DHP) exonsWe filtered 683,196 annotated human exons for those that show a threshold amount of variability in RNA-seq experiments from nine GTEx organ cohorts with between 226 and 653 samples each, identifying 141,043 exons that showed a degree of variable expression equal to or above a threshold of a mean count of at least 20 reads per sample and at least a two-fold ratio of the 95th percentile to the 5th percentile of expression values.
We classified the relationship between overall gene expression and the percent-spliced in (ψ) values of these exons, defining exons where increasing values of ψ (higher exon inclusion) are associated with higher gene expression as UHP exons (“upregulation of gene expression associated with high percent splice in”), exons where increasing values of ψ are associated with lower gene expression as DHP (“downregulation of gene expression associated with high percent splice in”), and exons that show alternative splicing without association between the ψ value and gene expression as type 0 exons (defined as a Benjamini–Hochberg-corrected p-value of at least 0.5). For each of the nine investigated tissues from the GTEx resource, we performed linear regression to predict gene expression based on ψ, and determined the significance of the coefficient for ψ. Raw p values were corrected for multiple testing by the Benjamini-Hochberg method, and associations are reported as significant at a corrected p-value threshold of 0.05 (Methods).
Using these heuristic definitions, we identified 3667 UHP and 3207 DHP exons; a total of 6874 unique exons were identified as UHP or DHP in at least one tissue, corresponding to 4.9% of the 141,043 exons that showed a at least a threshold level of gene expression variability (Methods). 989 exons were identified as UHP or DHP in multiple tissues (Fig. 1; additional examples are shown in Supplementary Fig. 1). In all, exons were identified as UHP or DHP 8282 times across the 9 tissues that were tested.
In all 989 cases in which exons were identified as UHP or DHP in multiple tissues, the assignment to UHP or DHP was consistent. We further used the same criteria to find the same UHP/DHP exons in sets of samples that originated from the same donor, for donors with at least 20 tissue samples. A total of 63,961 of the same UHP/DHP exons (1916 unique exons, ~89%) were detected in 528 donors. For 63,255 (~99%) the assignment to UHP or DHP was consistent with the assignment from tissue samples. The small number of inconsistencies is possibly a result of wrong classification due to the relatively small number of samples per donor (a median of 27 samples per donor vs. 342.5 per tissue).
We repeated the same analysis in unrelated breast, left ventricle and liver bulk RNA-seq datasets obtained from the SRA (Methods). In all three datasets, most of the overlapping exons were type 0 in both the GTEx and the SRA dataset, and most of the other exons were type 0 in one of the datasets. For the breast and left ventricle datasets, we observed a highly significant overlap of UHP or DHP classifications between the GTEx and SRA datasets. For liver, there were 52,521 exons that were classified as type 0, 17 exons that were classified as UHP and 47 exons that were classified as DHP. 14 exons were classified as DHP in both datasets, one exon was classified as UHP in both datasets, and all other exons were type 0 in at least one of the datasets (Supplementary Table 2). These results suggest that there is a significant consistency of exon types across different donor cohorts and experimental procedures.
Minimum prevalence of expression/splicing regulation couplingIn order to estimate how prevalent the coupling between expression and splicing is, we counted the number of exons that were neither detected as UHP nor as DHP, had a 95th/5th expression percentile ratio of at least 2, and were assigned a Benjamini-Hochberg-corrected p-value of at least 0.5, in addition to being expressed in at least half the samples in a tissue and at a mean level of 20 transcripts. This definition of type 0 exons intends to identify exons with substantial gene expression variability but with no evidence for being UHP or DHP exons. This resulted in 67,814 cassette exons identified as type 0. Since observing an effect of expression on splicing requires the presence of regulatory factors, such as RNA binding proteins, not observing a correlation does not immediately imply that an exon is type 0 in all tissues. However, since we examined nine different tissues, it is likely that there is roughly an order of magnitude difference between the counts of UHP/DHP exons and type 0 exons (6874 UHP/DHP vs. 67,814 type 0). In the nine tissue dataset from GTEx, there were a total of 8314 genes that contained at least one exon classified as UHP, DHP, or type 0. Of these, 1106 genes (13.3%) had at least one UHP or DHP exon. Supplementary Table 7 summarizes the number of UHP/DHP exons that were detected in the GTEx dataset, those that were detected in multiple tissues, and the overlap of these exons with exons detected in other datasets. While the number of UHP/DHP exons that are detected depends on the genes are expressed in each dataset, those genes vary in expression, statistical power and cellular mechanisms such as epigenetics modifications, the consistency in the direction of coupling suggests a core mechanism that if active, has a specific effect for each exon.
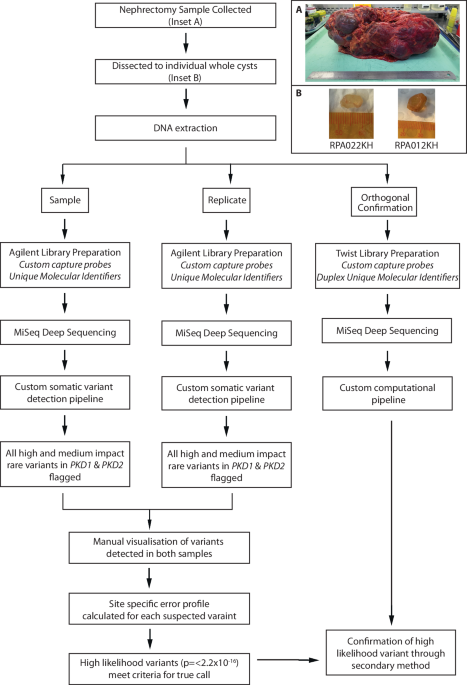
Characteristics of type 0, UHP, and DHP exons and the transcript and genes that contain themUHP/DHP exons differ from type 0 exons in a number of characteristics including exon count, intron length, and distribution of biotypes (Fig. 2). Genes containing UHP/DHP exons have on average more exons than genes containing only type 0 exons. The genes containing them had on average slightly fewer transcripts (13 and 12 for UHP and DHP, respectively, and 14 for type 0). Furthermore, type UHP/DHP exons are included in a larger proportion of transcripts than type 0 exons.
Fig. 2: Characteristics of type 0, UHP, and DHP exons.
a The number of exons of genes that includes a type 0, UHP, and DHP exon. b The number of transcripts per gene containing a UHP, DHP or type 0 exon. c Exon length in base pairs for type 0, UHP, and DHP exons. d The length of introns upstream of type 0, UHP, and DHP exons. e The length of introns downstream of type 0, UHP, and DHP exons. f Fraction of transcripts of each type associated with different biotypes. Green: protein coding; purple: retained intron; blue: protein coding CDS not defined; khaki: nonsense mediated decay; red: lncRNA. a–e Outliers were removed to limit the y-axis range; d–e The dashed red line shows the median for the up/downstream intron length type 0 exons. The figure was generated using ggpubr. Boxplot whisker lengths are the default (1.5 IQR).
We define the “upstream” intron as the last contiguous non-coding region that is transcribed 5′ to the exon, and the “downstream” intron as the first such region that is transcribed 3′ to the exon. The median upstream intron lengths were 572 bp for types 0, 857 bp for type UHP, and 732 bp for DHP; the differences between UHP or DHP and type 0 were statistically significant. In contrast, the median downstream intron lengths were 576 bp for type 0, 834.5 bp for UHP, and 485 bp for DHP. The differences are statistically significant between all types. DHP exons had a median length of 158 bp, which is significantly longer than UHP (median 135 bp) and type 0 (median 142 bp) exons. Finally, transcripts containing UHP/DHP exons have a higher fraction of protein coding transcripts (65% for UHP/DHP and 50.7% for type 0 exons), and a smaller fraction of retained introns (12.5% and 13% for UHP/DHP, respectively, and 20.5% for type 0) and long non-coding RNA (0.47%, 0.36% and 2.3% for UHP/DHP, and type 0, respectively) (Fig. 2 and Table 1). Additionally, the mean MaxEnt39 acceptor and donor splice site scores were higher for both UHP and DHP exons than for type 0 exons (Supplementary Figs. 3 and S4).
Table 1 Characteristics of type 0, UHP, and DHP exonsHigh consistency of UHP vs. DHP classification across multiple tissues and datasetsWe hypothesized that if the classification of exons as UHP or DHP is related to one or more core regulatory processes, then the classification should be largely conserved across different tissues. Among the detected UHP/DHP exons, there are 606 exons that appear in more than one tissue as DHP always, 383 that appear in more than one tissue always as UHP, and none that appear in more than one tissue as conflicting types. The slopes of the regression lines fitted in different tissues may have different slopes, but the change in slope is correlated across UHP/DHP exons (Supplementary Fig. 5). In addition, the slope is a linear function of the mean expression level, with coefficient close to 1, possibly indicating that differences in expression rates affect the impact of UHP/DHP exons on the gene’s transcript profile (Supplementary Fig. 2).
Distribution of RNA polymerase II binding in type 0, UHP, and DHP exonsRNA Pol II accumulates on exons in yeast and human and pauses over the 5′ and 3′ splice sites of human exons40. Additionally, Pol II density is lower at skipped exons than at alternative retained exons41,42. Based on the suggested mechanism (Fig. 1E), we hypothesized that RNAP2 density might differ between the type 0, UHP, and DHP exons investigated in the current study.
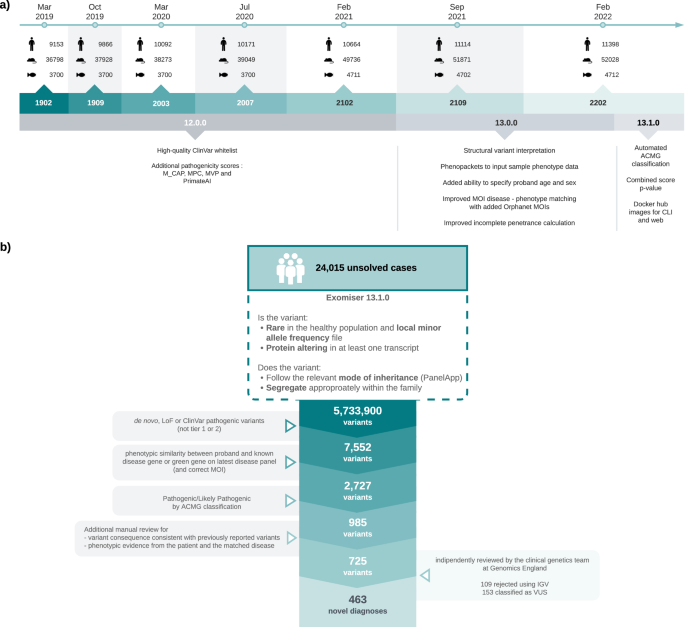
In order to estimate the difference in transcription speed of UHP and DHP exons compared to type 0 exons, we used two PRO-Seq datasets43,44 (Methods). These datasets sequenced nascent mRNA in addition to mature mRNA, and therefore allowed reads to be counted in the intronic parts of the nascent mRNA of each gene. The introns downstream of UHP/DHP exons are more likely to be sequenced, suggesting that RNA polymerase spends more time transcribing them (Fig. 3, Chi-squared test p < 2.2 × 10–308). The longer transcription time may be necessary for the regulatory interactions that promote or suppress the splicing of the exon, and thus may be sensitive to changes in expression rate. Supplementary Fig. 10 shows that RNAP binding to the exons themselves is likely to be lower for UHP/DHP.
Fig. 3: UHP/DHP exons and RNAP2 profiles.
a Intersection with PRO-Seq reads and downstream introns per exon type count, from the Wissink et al. dataset43 and the Gupta et al. dataset44. The dashed lines show the maximum difference between type 0 exons and other exon types that was obtained in 1000 permutations of the exon types (Fig. 3b). The values for UHP/DHP exons are significantly larger than type 0, suggesting a longer processing time of that section of the nascent mRNA. b Differences between the PRO-SEQ counts obtained for type 0 exons and the values obtained for other exon types, when the exon types for every hit are permuted 1000 times. The differences are much smaller than those obtained for the real data, suggesting that the results are statistically significant. The figure was generated using ggpubr. Boxplot whisker lengths are the default (1.5 IQR).
Enriched motifsBinding of transcription factors to promoters may influence splicing by altering the rate of RNAP2 elongation or recruiting splicing factors to pre-mRNAs45. We reasoned that if this were a common factor related to the mechanisms that underlie UHP/DHP exons, then we would expect to see enrichment of predicted transcription factor flexible model (TFFM) sites in the promoter regions of UHP/DHP exons compared to type 0 exons, and would also see enrichments of predicted RBP binding sites in the sequences surrounding the UHP and DHP exons. We therefore calculated the numbers of predicted binding sites and compared the observed counts to those observed in 1,000,000 permutations in which the labels of UHP, DHP, and type 0 exons had been randomly shuffled (Methods).
321 of 610 tested TFFMs showed significant enrichment in genes with UHP or DHP exons but no type 0 exons as compared to genes with at least one type 0 exon but no UHP/DHP exon. However, the maximum difference between the two classes was 3%, suggesting that no individual transcription factor is associated with a majority of the observed effects (Table 2, Supplementary Table 3). We tested enrichment for core promoter elements and CpG islands and found that a significantly higher proportion of DHP genes co-localized with a CpG island and a lower proportion contain a TATA box (Supplementary Table 4). We examined 71 RBP models, 31 of which showed significant differences between UHP or DHP and type 0 exons (Supplementary Table 5).
Table 2 Transcription factor flexible models (TFFMs) in promoters of genes harboring UHP and DHP exonsBiological rationale for coupling expression and splicingThe ubiquity of UHP/DHP exons led us to further investigate associations of transcript-specific functions. Alternative splicing of many genes can produce isoforms that differ with respect to enzymatic activities and subcellular localizations, as well as protein–protein, protein–DNA, and protein–ligand physical interactions46. Gene Ontology (GO) overrepresentation analysis is a standard approach to assessing the functional profile of differentially expressed genes47, but analogous methods for examining the functional profile of differential isoforms have not been available, possibly because of the paucity of experimentally confirmed functional annotations of isoforms48. We recently developed an expectation-maximization framework for predicting isoform-specific GO annotations49. We used these annotations to assess overrepresentation of GO terms in the set of isoforms that were found to be UHP in our study, with the universe of comparison being the set of all isoforms for which an exon of any type was detected. A total of 410 GO terms displayed significant overrepresentation (See Table 3 for the top ten and Supplementary File 2 for a complete list).
Table 3 The ten GO terms in which UHP isoforms are most over-representedInterestingly, the top three overrepresented terms, JUN kinase activity (Supplementary Fig. 6), MAP kinase activity, and MAP kinase kinase activity annotate genes and pathways that coordinately regulate gene expression, mitosis, metabolism, motility, survival, apoptosis, differentiation and protection against DNA damage, and deleterious mutations50,51,52. Our results suggest that cells can upregulate these pathways both by increasing overall expression of genes in the pathway and also by alternative splicing to favor transcription of isoforms that specifically possess pathway activity.
We investigated the association between the degree of differential expression and the degree of alternative splicing by regressing ψ against the gene expression level for all exons identified as UHP/DHP. We found that the slope of the expression-ψ line is approximately the mean expression level of the gene (Supplementary Fig. 2). Therefore, in dividing cells, which need to double their protein content, increasing expression of genes annotated to JUN kinase activity, MAP kinase activity, and MAP kinase kinase activity will additionally strongly favor transcription of UHPs that also are specifically annotated to these GO terms. Thus, increased gene expression will both increase the overall amount of genes and shift their transcript distributions to transcripts with Jun kinase activity. In order to test this hypothesis, we computed the Pearson correlation between UHP exons \(\psi \) and Cyclin D1 gene expression, as the latter is a marker for the level of mitosis53. This correlation is mostly positive, while the same correlation between DHP/Type 0 exon \(\psi \) and Cyclin D1 expression is mostly negative (Supplementary Fig. 7). We further calculated correlation between UHP exons \(\psi \) and Cyclin D1 expression in The Cancer Genome Atlas (TCGA) transcript expression dataset, for primary tumor and normal tissue, and found a significant reduction in correlation in tumor samples (Supplementary Fig. 8). Synchronization of alternative splicing with the cell cycle has been previously observed by Domingues et al. 54. Their list of genes significantly overlaps with genes that contain UHP or DHP exons (p-value 0.01, Hypergeometric Test). Schor et al. also suggested that the coupling mechanism may be altered in cancer55. Based on our findings, we suggest a potential mechanism by which the degradation of expression-splicing coupling may contribute to cancer development. At early stages of cancer development, when the cells divide quickly but have not accumulated a large number of mutations, the coupling is intact, and might even be activated compared to normal cells if the normal cells divide slowly. At later stages, as more mutations accumulate, the coupling is reduced as splicing factor binding sites and splicing factors are affected by mutations. Since this does not prevent the cells from further dividing, we speculate that the pathways that are no longer induced by the cell cycle are intended to prevent genomic instability, for example through the inclusion of transcripts in the Jun Kinase and MAP Kinases pathways. In order to test this idea, we separated the cancer samples in Supplementary Fig. 8 to those that had less than 20 non-silent mutations (SNPs and indel) and those that had 20 or more. As can be seen in Supplementary Fig. 9, the correlation of UHP PSI with Cyclin D1 expression decreased with increase in the number of SNVs, which supports the hypothesis that the coupling degrades gradually.
Many of the top enriched terms of DHP-containing transcripts are involved in monosaccharide metabolism, e.g., glycolysis, fructose metabolism, glycolysis from storage polysaccharide and glycogen catabolism (Supplementary File 3 contains a complete list). We speculate that this coupling could promote efficient replenishment of energy reserves after cell division or generate energy for future division. It is reasonable to assume that DHP exons are also used to shut down pathways whose activity is undesirable during cell division. We defer a focused analysis of the functional synergy between UHP and DHP exons to a future study.
留言 (0)