
記住我



This is a cross-sectional study utilizing data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet), funded by the Centers for Disease Control and Prevention (CDC), a network that conducts population-based surveillance, longitudinal follow-up, and public health research about muscular dystrophies. The individuals identified during the surveillance period (January 1, 2008- December 31, 2016) had clinical records of varying lengths; some individuals were followed for a few visits, and others had many years of data available (see Table 1a, 1b). The analysis focuses on symptoms of each patient reported up until their last available medical provider visit.
Table 1a Categorical characteristics by sex, among adult-onset DM1 participants from MD STARnet (n = 228)Table 1b Continuous characteristics by sex, among adult-onset DM1 participants from MD STARnetThe surveillance sites were the U.S. states of Colorado, Iowa, 31 counties in North Carolina’s Piedmont region, South Carolina, Utah, and a 21-county area in Western New-York. Eligible individuals in these catchment areas were residents who had a clinic visit for DM1 during the eight-year study period. Earlier versions of the MD STARnet methodology are described in detail elsewhere [25, 26]. Here we briefly describe the methodology for the data used in this analysis.
MD STARnet used multi-source case finding with a focus on neuromuscular clinics. Other data sources included hospitals and hospital discharge databases, private physician practices, service sites for children with special health care needs, and birth defects surveillance programs. Identified individuals were linked to birth and death certificate data for more complete ascertainment of deaths. Potential DM1 individuals were identified by International Classification of Diseases, Ninth or Tenth Revision, Clinical Modification (ICD-9-CM or ICD-10-CM) codes (359.21, myotonic muscular dystrophy; G71.1, myotonic dystrophy; G71.0, muscular dystrophy) in the electronic medical records of specialty clinics, as well as death records.
Trained abstractors who were blinded to the study hypotheses screened records for demographic and clinical information. Two to three abstractors per MD STARnet site were trained in clinical aspects of DM and each site had a supervisor who did clinical review of the abstracted records. A MD STARnet database was used to code detailed diagnostic information, including S/S, initial history, physical examination, diagnostic notes, clinical progress notes, family history, electromyography reports (EMG), muscle biopsies, and genetic laboratory reports. Additional information, without a field, was copied verbatim from the clinical record into the database. Following data collection, a committee of seven neuromuscular clinicians, one neurologist or neuromuscular specialist per MD STARnet site, reviewed the abstracted diagnostic data to assign each individual a case status [27]. A definite case has myotonic dystrophy related clinical symptoms and genetically confirmed diagnosis by a clinical blood test (19q13.3 CTG repeat length > 50) either from the patient or from the family member. A probable case has related clinical symptoms and supported by family history of first-degree family member (either maternal or paternal transmission) consistent with DM1. Asymptomatic cases had a confirmed DNA diagnosis (same as Definite), but none of the following clinical symptoms: for children: weakness, myotonia (clinical or electrical); for adults: development of cataracts before age 50, myotonia (clinical or electrical), daytime hypersomnolence, or distal or proximal weakness.
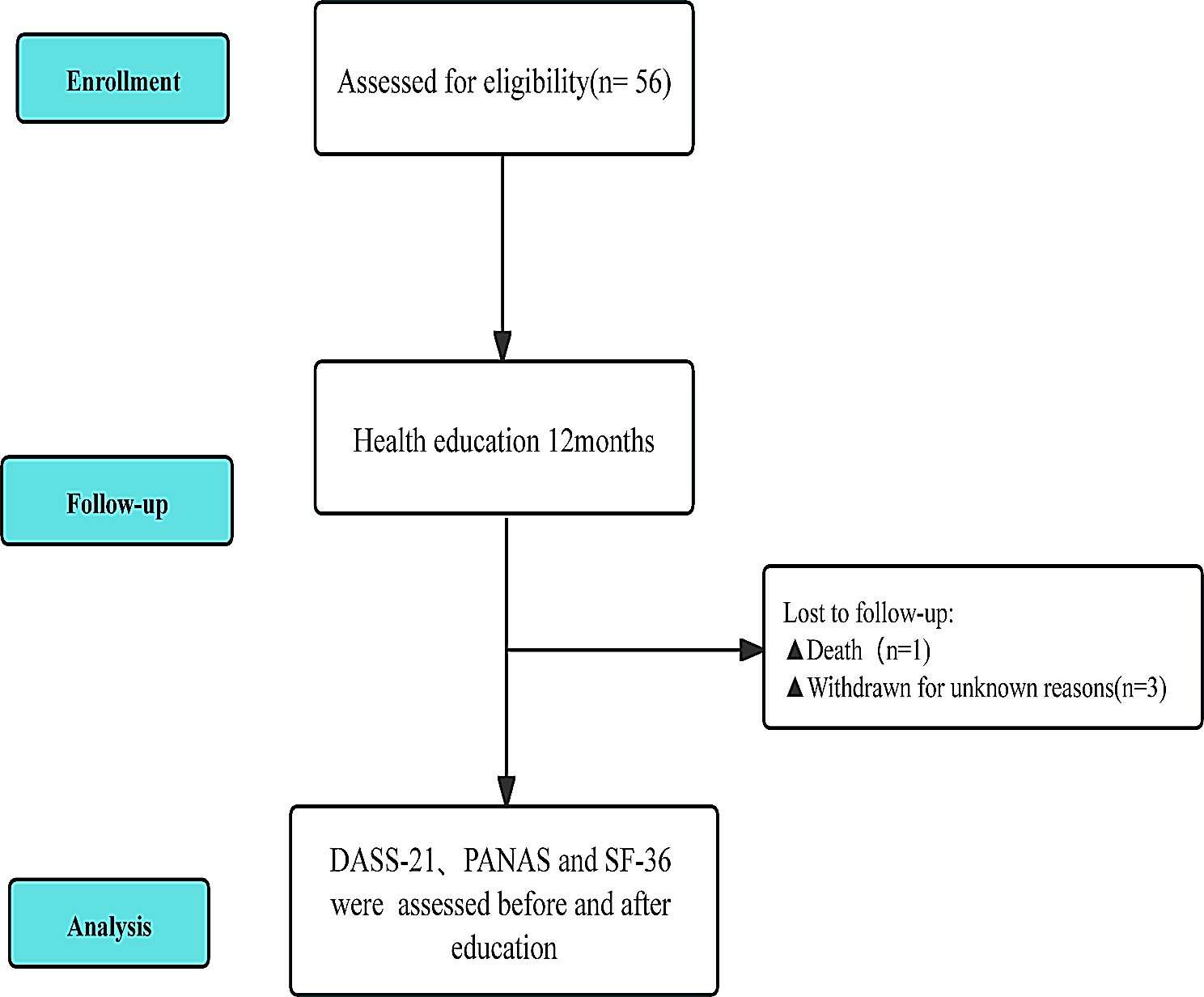
Study sample inclusion criteriaInclusion and exclusion criteria for this study are displayed in Fig. 1. There were 486 individuals ascertained with myotonic dystrophy in MD STARnet during the period 2008–2016. Considering the differences in gene mutations, age of onset, as well as clinical presentations, we included only individuals diagnosed with DM1 and with a definite or probable case status. We also excluded four individuals who resided in Nevada due to concerns of inconsistent case collection; 24 individuals because their S/S are not consistent with medical records, and one individual who had no record of S/S. We chose to study adult-onset DM1 since three quarters of people with DM1 do not report S/S until they are older than ten years, and congenital and early childhood DM1 have different features in their presentation and a different trajectory of disease progression [16]. The final study sample was 228 individuals with onset after age 10 years.
Fig. 1
Flow chart of identified cases among adult-onset DM1 participants from MD STARnet. Based on the inclusion and exclusion criteria, a total of 228 individuals with adult-onset DM1 were included in the final analysis
OutcomesThe S/S in all clinical records were extracted, beginning with disease history, symptoms recorded during the initial contact with a neurologist or neuromuscular clinic, and S/S identified throughout subsequent visits. After grouping synonyms (Supplementary Table 2), twenty-one core clinical S/S were identified. While symptoms may be identified in multiple medical records, they are not analyzed as repeated measures. Regardless of when the symptoms were presented, the surveillance methodology only allowed for recording of symptoms until the patient’s last available medical provider visit.
Predictors /covariatesWe considered two primary independent variables: sex (male and female) and CTG repeat length (number of repeats of a CTG triplet in the 3’ non-coding region of DMPK). We classified CTG repeat length into the following categories: [(50–150), (150–500), (500–1000), and ≥ 1000]. For individuals who didn’t have genetic test results, their CTG repeat length was coded as ‘Not available’. We also considered two potential confounders, race/ethnicity and family history. Due to small case counts in some categories, we combined race and ethnicity into non-Hispanic White and other (including Hispanic, non-Hispanic across multiple races, and unknown ethnicity). Based on the family history in the medical records, family history was categorized into ‘Yes’, ‘No’ and ‘Not available’. Age of onset was defined as the age of the earliest reported S/S. Age at genetic test was defined as the age when the first genetic test was done. The length of follow-up was measured from the date of onset to the last available date of medical provider’s visit.
Statistical analysisCategorical characteristics were described by their frequencies and percentages, and their differences were assessed by ?2 or Fishers Exact test for small cell sizes. The continuous characteristics were described by mean and standard deviation or median and interquartile range (IQR), and their differences were tested by t-test or Wilcoxon rank sum test when normality was violated.
We first used exploratory factor analysis (EFA) to group the 19 S/S listed in Table 2. S/S reported by less than 10 individuals (respiratory failure/insufficiency, developmental delays) were considered rare and were not included in the factor analysis (pairwise deletion). Considering the convention that requires 10–15 cases per item, and with 19 items (S/S) in our study, the sample size (n = 228) was sufficiently powered for EFA. We estimated the polychoric correlation matrix, and we applied oblimin rotation by assuming factors are correlated since S/S are rarely partitioned into neatly independent factors. Weighted least squares adjusted to mean and variance was used as the estimator. The number of factors was decided by the combination of scree plot and model fitting statistics (Supplementary Table 1). A cutoff point of 0.3 was used to decide which S/S were included in a factor [28]. Myalgia had loadings less than 0.3 and were removed, and the EFA was refit. For S/S with cross-loadings greater than 0.3, we classified the S/S into the factor with the higher loading.
Table 2 Number of adult-onset DM1 individuals with each sign and symptom, from MD STARnet (N = 228)We then conducted a confirmatory factor analysis (CFA). Three statistically non-significant S/S (jaw stiffness p = 0.688, cognitive impairment p = 0.332, and cataracts before 50 for those over the age of 50 years p = 0.101) were excluded from further analysis. Individuals were assigned a factor score, which was computed by a count of S/Ss and their loadings. The associations of factor scores (outcome) with the logarithm of CTG repeats, age of onset, sex, race/ethnicity, family history status, years of follow-up since onset, and MD STARnet surveillance site were explored by fitting a linear regression model. Given the sample size and dimension of symptoms, we fit EFA and CFA using the same dataset, which could result in overfitting.
We assumed the missing data were non-informative for age of onset (n = 58, 25%), CTG repeat length (n = 68, 30%) and family history (n = 4, 1.8%). The sample size reduced from 228 to 115 when fitting the linear regression. All tests were two-tailed, and type I error probability was set at 0.05. For multiple linear regression, the false discovery rate was controlled using the Benjamini-Hochberg procedure. Specifically, we fit three separate multiple linear regressions examining the significance of six covariates, resulting in a total of 18 tests. The Benjamini-Hochberg procedure was applied by ranking the 18 p-values in ascending order. Each p-value was compared to a threshold of 0.05*rank/18 (e.g., the smallest to 0.05/18, the second smallest to 0.05*2/18, the next smallest to 0.05*3/18, and so forth, up to the largest p-value compared to 0.05). EFA and CFA were done using Mplus and R software. Descriptive statistics and linear regressions were done using SAS software 9.4 (SAS Institute Inc). All analyses were replicated by a second analyst.
留言 (0)