
記住我



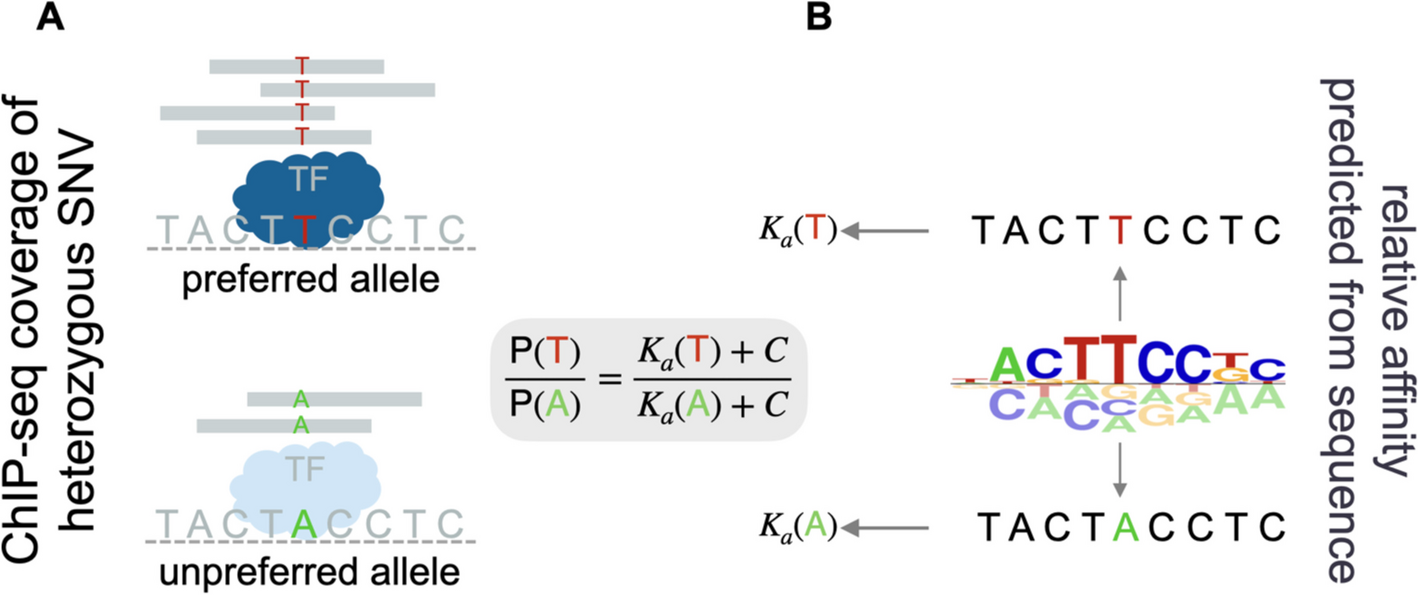






To assess to what extent sequence-to-affinity models can predict allele-specific binding effects, we used human ASB annotations from AlleleDB [5] and predicted the SNV’s effect on TF binding affinity. We chose to focus on the insulator protein CTCF due to the abundance of variants (2231 SNVs) with statistically significant evidence of ASB for this factor [5]. The MotifCentral.org database contains hundreds of ProBound models trained on in vitro binding data from HT-SELEX [12] and SMiLE-seq [13] assays. We used the MotifCentral model for CTCF to predict allele specific binding at heterozygous SNV loci that were previously found to have significant allelic bias in ChIP-seq coverage (Fig. 1A). For each SNV observed to have ASB, we predicted cumulative CTCF binding affinity from the DNA sequence around the SNV by summing over all offsets of the TF-DNA binding interface relative to the SNV, separately for the reference and the alternative allele (Fig. 1B; see “Methods” for details).
Fig. 1
Overview of the allele-specific binding data and binding affinity scoring. A For each variant, the preferred and unpreferred allele are defined in terms of ChIP-seq read coverage. B The binding affinity for each variant is computed as the sum of relative affinity scores over all possible offsets
A key question is to what degree the direction of the in vivo allelic imbalance in TF binding is concordant with the difference in the binding affinity as predicted by the sequence-to-affinity model. We used the MotifCentral model for CTCF as derived from in vitro binding data (Fig. 2A) to predict the direction of allelic preference from DNA sequence alone for each ASB variant (Fig. 2B). The overall concordance without regard of the precise numerical value of the respective affinity for each allele, only the direction of the difference, was 62.4%. While this is significantly larger than the expected value of 50% in the random case (binomial test p-value < 10−16), it is too far below 100% to be practically useful. However, two sequence-derived metrics can be used to stratify variants in terms of the likelihood that their allelic preference will be correctly predicted: First, we found that a large fold-difference in predicted affinity is associated with much higher concordance (Fig. 2C). Second, the predicted affinity of the strongest allele is informative in a complementary way, with substantial improvement in concordance seen over a remarkable thousand-fold range in affinity before it finally deteriorates (Fig. 2D); the latter may reflect both that the predictions of our model become less accurate at lower affinities and that the affinity-mediated effect of the variant on TF occupancy gets smaller compared to other contributions. When we imposed lower bounds on both of these metrics simultaneously, even higher levels of concordance can be achieved (Fig. 2E). Similar trends (Additional file 1: Figure S1) were seen for the only two other TFs represented in AlleleDB that have at least 100 variants with evidence for ASB (387 variants for PU.1, and 189 for EBF1). Taken together, these results point to the feasibility of sequence-based prediction of ASB via quantification of the difference in binding affinity between the two alleles, provided that it is possible to assess the reliability of such affinity predictions.
Fig. 2
Predicting allele-specific binding by CTCF using a sequence-to-affinity model. A Energy logo [22] representation of a CTCF binding model from MotifCentral capable of making accurate predictions of binding affinity. B Scatterplot of CTCF single-nucleotide variants (SNVs) at which significant ASB was detected in the AlleleDB study [5], colored by concordance. The x-value corresponds to the greater of the predicted affinities of the two alleles; the y-value corresponds to the ratio of predicted affinities of the two alleles. C, D Concordance of allelic preference, in bins of 100 SNVs as ranked by either of the axes in B. E Two-dimensional cumulative concordance of allelic preference as in B for all cutoffs with 20 or more SNVs
A metric for TF binding model quality based on allelic-specific ChIP-seq counts across the genomeSince for most TFs the fraction of variants for which allele-specific binding can be demonstrated on an individual basis is very small, we set out to find a way to aggregate below-threshold evidence for allelic preferences across the genome. We settled on a likelihood framework that uses the same beta-binomial distribution used by [5] to model allelic counts for AlleleDB; however, rather than using this model to reject the null hypothesis that both alleles of a given variant are equally probable, we used the likelihood function to quantify the performance of sequence-based predictors of allelic preference on a genome-wide scale (see “Methods” for details). Importantly, this was done without regard to the statistical significance of allelic imbalance at the level of individual variants.
AlleleDB [5] used a beta-binomial test with a probability of 1/2 for both alleles to detect instances of ASB from allele-aware ChIP-seq data. By contrast, we here used allele-specific ChIP-seq counts aggregated across multiple individuals, and re-estimated the overdispersion parameter underlying the distribution by maximizing the likelihood function. We reasoned that using a beta-binomial model with variant-specific allelic probabilities based on predicted CTCF binding affinities should improve the likelihood, compared to the equal-preference control.
We found that this was indeed the case for the sequence-to-affinity model for CTCF in MotifCentral. To assess the statistical significance of the difference, we used bootstrapping to sample the log-likelihood distribution for each binding model separately (Fig. 3; see “Methods” for details). Compared to the equal-preference control, the MotifCentral model has a significantly higher mean log-likelihood (− 3.2356 for control, − 3.2089 for MotifCentral; Wilcoxon test p < 10−8). Consistently, the maximum-likelihood estimate of the overdispersion parameter is lower (0.0916 for MotifCentral, 0.0990 for control). As expected, including all possible offsets between the binding model and the variant is important when predicting cumulative affinities from its flanking DNA sequence (Additional file 1: Figure S2). We saw modest further improvement in the likelihood when including contributions from offsets at which there was no overlap between the scored sequence and the variant, consistent with an interpretation that additional binding sites in the flanks would blunt the difference between the alleles when their effects on ChIP enrichment are additive.
Fig. 3
Bootstrap distributions of log-likelihood for CTCF. The histograms show the bootstrap distributions of log-likelihood for 1000 resamples, based on the affinity-based likelihood model (allelic ratio predicted from genome sequence using binding model) or the control model (which assumes the alleles are equiprobable). The vertical line indicates the observed value of log-likelihood from each model
We applied the same overall analysis to PU.1 and EBF1. Again, the MotifCentral model had statistically significant predictive performance when aggregating evidence across all variants (Additional file 1: Figure S3). Overall, our likelihood-based framework for integrating evidence across the genome based on the beta-binomial distribution provides a way to quantitatively compare sequence-based predictors of allelic preference in an aggregate manner that is not limited by the number of variants that reaches statistical significance for calling ASB at the individual-locus level.
In vivo datasets can be affected by DNA fragmentation bias and overly short fragment lengthsSo far, the TF binding models we used to score binding affinity were all derived from in vitro SELEX data. We wondered whether models derived in an allele-agnostic manner from in vivo assays would be better at explaining the allelic imbalances reflected in the allele-aware ChIP-seq data. To this end, we configured PyProBound to analyze paired-end in vivo data without peak calling (see “Methods” for details). Notably, when using the ChIP-seq data for CTCF from ENCODE, we found that the length of the merged read pairs was large enough for PyProBound to infer accurate binding models without any mapping to the genome.
Initially, when using PyProBound to fit a single binding mode intended to represent the DNA binding specificity of CTCF, the associated positional profile also learned by PyProBound indicated a strong bias at the ends of the ChIP-seq fragments (Additional file 1: Figure S4A). We interpreted this as confounding between the sequence preferences of CTCF binding and an unknown local sequence dependence of DNA fragmentation at the ends of the paired reads. To address this, we configured PyProBound to fit a more complex model that accounts for two multiplicative effects simultaneously: (i) the sequence dependence of the rate of DNA fragmentation during sonication at the two observed ends of the fragment; and (ii) the sequence dependence of CTCF binding, which determines the probability that the fragment is crosslinked with this TF during immunoprecipitation (see “Methods” for details).
This more sophisticated use of PyProBound when learning a sequence-to-affinity model from allele-agnostic ChIP-seq data for CTCF led to significantly improved performance when predicting ChIP-seq enrichment across the genome, and made the positional bias disappear (Additional file 1: Figure S4C). Note that a simpler approach in which we truncated each fragment to 200 bp around its center, which obscures the fragmentation bias due to the varying length of the fragments, performed less well on predicting enrichment during the immunoprecipitation step (Additional file 1: Figure S4B).
We found that PyProBound analysis of CUT&Tag and ChIP-exo data for CTCF did not require explicit fragmentation modeling, which allowed us to add additional flanking sequence to the genomic fragments defined by mapped read pairs to account for additional binding sites contributing to the enrichment achieved by the assay, improving the ProBound loss (from 1.5555 to 1.5308 for CUT&Tag after extending by 1300 bp, and from 1.3939 to 1.3917 for ChIP-exo after extending by 200 bp).
Taking DNA fragmentation bias into account when analyzing ChIP-seq data yields superior sequence-to-affinity modelsTo summarize the performance of the various alternative sequence-to-affinity models for CTCF thus generated, we again used our ASB likelihood metric (Fig. 4). The results were consistent with expectation in various regards: (Py)ProBound models trained on in vivo binding data in general outperform those trained on in vitro data. Additionally, ChIP-seq-derived PyProBound models trained directly on fragments outperform those trained on sequences extracted from genome alignments, as well as models trained on fragments that were all truncated to be the same length (log-likelihood −3.1989 for raw fragments; −3.2089 for length truncated; −3.2016 for genome alignment). CUT&Tag and ChIP-exo models improve in performance when trained with additional flanking sequences (log-likelihood improved from −3.1992 to −3.1963 for CUT&Tag after extending by 1300 bp, and from −3.1988 to −3.1979 for ChIP-exo after extending by 200 bp). Moreover, the PyProBound model inferred from CUT&Tag data showed better performance than either the PyProBound model inferred from ChIP-seq or ChIP-exo data or the motifs for CTCF available from JASPAR and HOCOMOCO (Fig. 4). Comparing the models on the less principled but perhaps more intuitive coefficient of determination (Pearson R2) of binned allelic ratio yielded the same ranking in performance (Additional file 1: Figure S5).
Fig. 4
Comparison between bootstrap distributions of log-likelihood across various models for CTCF binding. The boxplot shows the comparison in log-likelihood between the control model and affinity-based models based on different CTCF binding motifs, including a CUT&Tag-derived model introduced in this study and motifs from various resources
Since models from external resources may not be well-equipped to predict relative binding affinities, we also wished to compare them on the simpler task of predicting only the direction of allelic preference, to investigate why they underperform on the ASB likelihood metric. Using the two metrics discussed above of predicted score fold-difference and predicted score of the strongest allele, we found that HOCOMOCO and JASPAR models of CTCF binding systematically underperform compared to MotifCentral in terms of allelic preference concordance; the performance gap is strongest among the sequences that these models rank as the highest predicted binders (Additional file 1: Figure S6).
De novo motif discovery from allele-specific ChIP-seq countsOur success in predicting allelic preference on a genome-wide scale using independently derived TF binding specificity models suggests that the direct modulation of TF binding by variants can explain allelic imbalance to a significant degree. We therefore wondered whether the same beta-binomial likelihood function could be leveraged to perform de novo motif discovery purely by trying to explain allelic imbalances in ChIP-seq counts from DNA sequence, while taking variation in combined ChIP-seq coverage among variants for granted. To our knowledge, such an approach, which controls for variation in chromatin context in a unique way by implicitly assuming a similar local molecular environment of a given variant on the respective homologs of the chromosome on which it resides, has not been explored before. We configured PyProBound to optimize the beta-binomial likelihood function not only with respect to the overdispersion parameter and non-specific binding coefficient as was done above, but also with respect to the position-specific free-energy parameters of the TF binding model (see “Methods”). Since we could not compare the resulting model on the benchmark data it was trained on, we instead compared its energetic parameters against previously published models (Fig. 5A). The ASB-derived model showed excellent agreement with the MotifCentral model (Pearson R2 = 0.890; Fig. 5B), indicating that CTCF allelic imbalance is indeed driven by direct alteration of sequence-specific CTCF binding. We used the same motif discovery approach for EBF1, which resulted in a model that again had good agreement with the corresponding MotifCentral model (Pearson R2 = 0.817; Additional file 1: Figure S7A). For PU.1, however, the ASB-derived model discovered additional specificity at one end of the binding mode compared to the MotifCentral model, leading to inferior agreement (Pearson R2 = 0.739; Additional file 1: Figure S7B). A wider binding model capturing this additional specificity instead best compared to the HOCOMO model for PU.1 (Pearson R2 = 0.823; Additional file 1: Figure S7C), indicating that a difference in in vitro and in vivo binding specificity for PU.1 may underlie these observations.
Fig. 5
Comparison of de novo ASB-derived and existing MotifCentral CTCF models. A Energy logos for binding model inferred from allele-aware CTCF ChIP-seq data from [5] using PyProBound, and from CTCF SELEX data using the original version of ProBound [18]. B Direct comparison of free energy parameters in the respective models, with each point corresponding a unique base/position combination in the logo
留言 (0)