
記住我
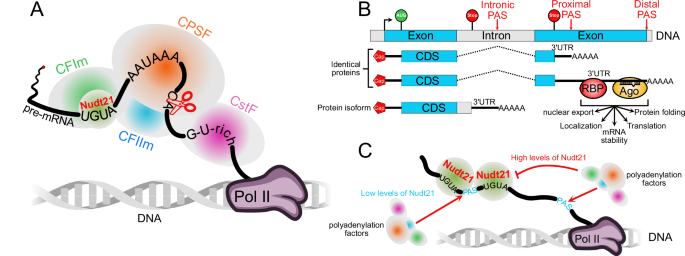
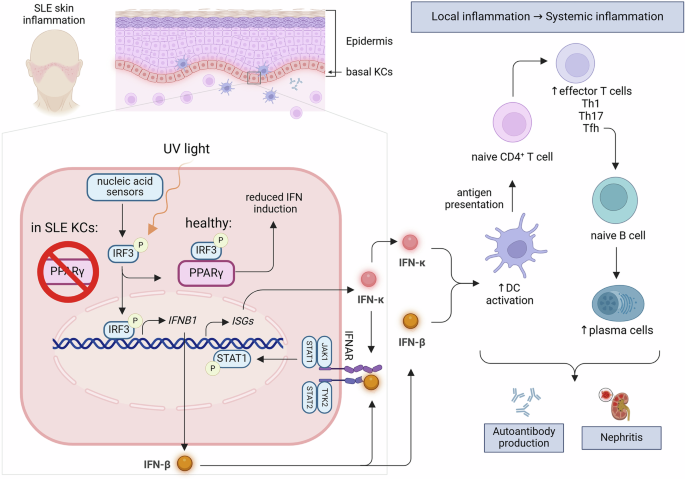
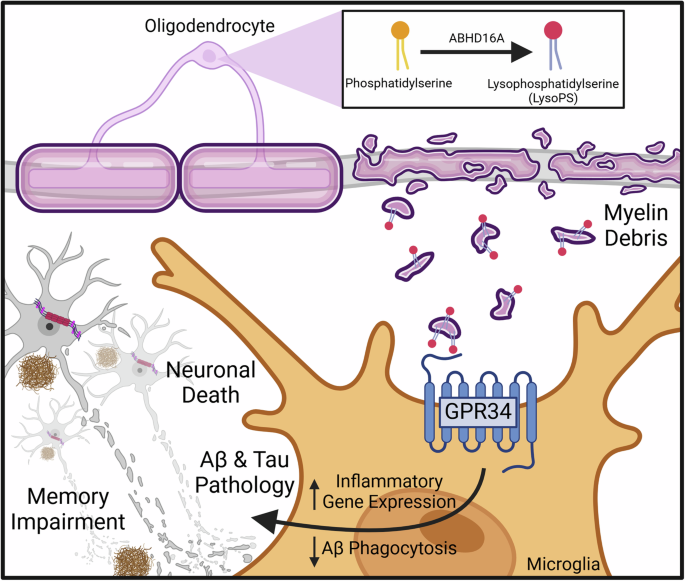
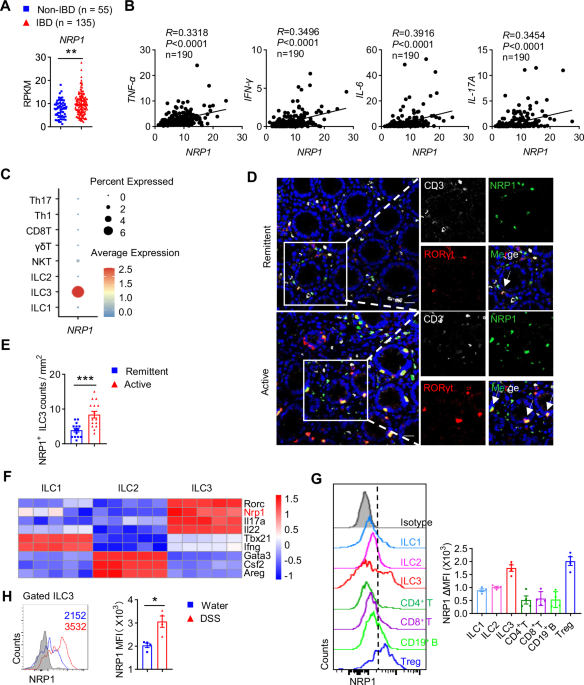
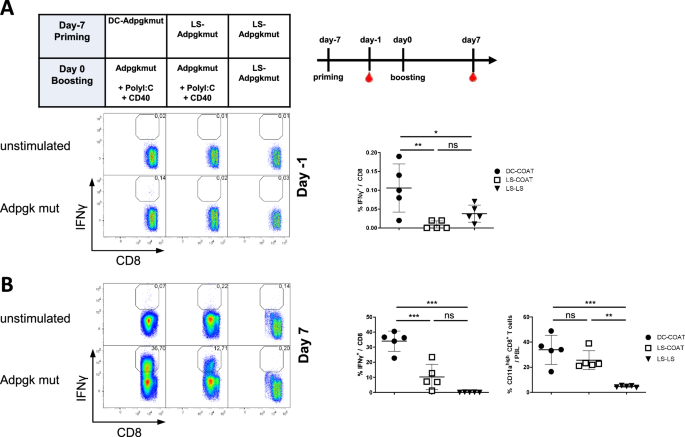

Twelve patients with sepsis or septic shock were included in the study. Community-acquired pneumonia was the most common cause in this study, accounting for eight cases (66.7%). Supplementary Table 2 summarizes the patients’ demographic and laboratory characteristics. There were four septic shock patients (33.3%). Eight patients (66.7%) required mechanical ventilation, and one had a tracheostomy. The 28-day mortality rate was 25%. Eight patients had comorbidities, six of whom had more than two. The comorbidities had no significant effect on ICU mortality. Eight patients (66.7%) had causative pathogens, with Escherichia coli being the most common. Supplementary Table 3 compares the characteristics of patients who recovered within 48 h to those of patients who did not recover within 48 h of septic shock. Univariate analysis revealed significant differences in survival rates between the two groups.
Extraction of RNA or mitochondrial DNA (mtDNA) contentsTotal RNA was extracted via TRIzol reagent (Invitrogen, Waltham, MA, USA, 15596026) according to the manufacturer’s instructions. cDNA synthesis was carried out via reverse transcriptase premix (ELPIS Biotech, Daejeon, South Korea, EBT-1515) following the manufacturer’s instructions. To determine the mtDNA copy number, total DNA was extracted from BMDMs via a DNeasy blood and tissue kit (Qiagen, Hilden, Germany, 69504) following previous methods [91, 92].
Bulk RNA-seq of human-derived materialsAfter total RNA was extracted from each sample, the mRNA was enriched with oligo(dT) magnetic beads, and the rRNA was removed. After the addition of fragmentation buffer, the mRNA was fragmented into short fragments (approximately 200 bp), and the first-strand cDNA was subsequently synthesized via random hexamer primers with the mRNA fragments used as templates. Buffer, dNTPs, RNase H, and DNA polymerase I were added to synthesize the second strand. The double-strand cDNA was purified with a QIAquick Gel Extraction Kit & PCR Purification Kit (Qiagen, 28704 and 28104, respectively) and washed with EB buffer (elution buffer; Qiagen, 19086) for end repair and poly(A) addition. Finally, sequencing adapters were ligated to the fragments. The required fragments were purified via agarose gel electrophoresis and enriched via PCR amplification. The library products were assessed with a BioAnalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). The resulting cDNA libraries were sequenced via the HiSeq 4000 platform (Illumina, San Diego, CA, USA), generating approximately 1.44 billion paired-end reads of 151 nucleotides in length. The raw RNA-seq data have been deposited in the NCBI SRA database with the following accession numbers: SRX8138400--SRX8138419 under BioProject PRJNA625581 and SRR28762191--SRR28762194 under BioProject PRJNA1102979.
Bulk RNA-seq analysis of human-derived materialsTo obtain high-quality clean reads for transcript analysis, all raw sequence reads were preprocessed via Trimmomatic (v0.36) [93] to trim the adapter sequences and remove low-quality sequences. The remaining clean reads for each sample were aligned independently to the human reference genome (hg38) via HISAT2 (v2.1.0) [94]. To assemble and quantify the transcripts, the resulting aligned reads and human annotation data were input into Cufflinks (v2.2.1) [95]. We subsequently merged the transcriptome assemblies from each sample via the Cuffmerge script (v2.2.1) implemented in Cufflinks and applied Cuffdiff (v2.2.1) [96] with default parameters for the identification of DEGs. In this study, we defined genes as differentially expressed genes with an FDR of less than 5%. The human reference genome and annotation data were obtained from the UCSC genome browser (https://genome.ucsc.edu), and the data for visualization were generated via R (R Development Core Team, Vienna, Austria). Unless otherwise stated, the unit of expression level in our analysis was fragments per kilobase of exon per million fragments mapped (FPKM). Gene Ontology (GO) enrichment analysis was performed with the DAVID (v6.8) functional annotation analysis tool (https://david.ncifcrf.gov) [97].
Single-cell RNA-seq (scRNA-seq)We performed library construction via 10× Chromium Single Cell 3’ reagent kits v3.1 and sequenced the libraries on the NovaSeq 6000 platform (Illumina). The initial sequencing data were processed and converted into FASTQ files via the Cell Ranger pipeline. We adhered to the standard sequence protocol recommended by 10× Genomics, which involves trimming the barcode and unique molecular identifier (UMI) ends at 26 base pairs and the mRNA ends at 98 base pairs. Following this preprocessing step, the resulting FASTQ files were aligned to the human reference genome (GRCh38). We subsequently employed Cell Ranger for preliminary data analysis, resulting in the generation of a data file comprising a barcode table, a gene table, and a gene expression matrix. To analyze the scRNA-seq data, we utilized R with Seurat (v.4.0.5) [98] to process the single-cell read counts obtained from each sample. To ensure data quality, we filtered out cells on the basis of the following criteria: cells with UMI counts per cell less than 500, cells with genes detected per cell less than 300, and cells with a mitochondria ratio exceeding 10%. Subsequently, filtered Seurat objects were integrated, and data normalization was performed via SCTransform to correct for batch effects originating from different samples. Next, highly variable genes were identified via the FindVariableFeatures function within Seurat. These identified genes were then subjected to principal component analysis for linear dimension reduction. Subsequently, cell clusters were identified via the FindClusters function, with the resolution parameter set to 0.05. To visualize the cell clusters, we employed t-distributed stochastic neighbor embedding (tSNE). The scRNA-seq data and analysis script are available at https://github.com/tjdrnjsqpf/UBXN6_scRNA-seq.
MiceThe mice used in individual experiments were age- (6–8 weeks) and sex-matched. Ubxn6flox/flox mice were purchased from Cyagen Biosciences (Jiangsu, China; CKOCMP-66530-Ubxn6-B6J-VA). Lys2Cre mice were kindly provided by Dr. C.-H. Lee (Korea Research Institute of Bioscience and Biotechnology). The mice were maintained under specific pathogen-free conditions. The animal experiments and handling were performed following the ethical guidelines of the Chungnam National University College of Medicine and were approved by the Institutional Animal Care and Use Committee (202109A-CNU-180; Daejeon, South Korea) and the South Korean Food and Drug Administration.
GenotypingHomozygous targeted mice from heterozygous breeding pairs were generated. Semiquantitative PCR was conducted by using Power S Taq Premix (HKGenomics, Daejeon, South Korea, 11201). The tissue-specific gene deletion was confirmed by the following primer sequences (5 to 3 primers): Ubxn6 conditional wild-type (Ubxn6 cWT); forward, 5-GAG GAA CAT GGA GGT TCA AAG GA-3; reverse, 5- CAG TGC AGT TCA GAG GCA GGT T-3. Ubxn6 conditional knockout (Ubxn6 cKO); forward, 5-GAG GAA CAT GGA GGT TCA AAG GA-3; reverse, 5- AAG TCT CGT GTT GAA CTC CTT ACA-3. The fragment size was 431 bp for the Ubxn6 cWT allele and 363 bp for the Ubxn6 cKO allele.
Isolation, cultivation, and treatment of cellsHuman PBMCs were isolated from heparinized venous blood by density sedimentation over Ficoll-Hypaque (Lymphoprep; Alere Technologies, Oslo, Norway, 07851). The cells were incubated for 1 h at 37 °C, and nonadherent cells were removed by pipetting off the supernatant. Adherent monocytes were then resuspended in Roswell Park Memorial Institute 1640 medium (Corning, 1 Riverfront Plaza, NY, USA, 10-040-CVRC) containing 5% human serum (Sigma‒Aldrich, St. Louis, MO, USA, H3667) and 1% L-glutamine. Primary BMDMs were obtained as follows. Bone marrow cells were harvested from the femurs and tibias of 6–8-week-old mice and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Lonza, Walkersville, MD, USA, BE12–604 F) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA, 16000–044) and a penicillin‒streptomycin‒amphotericin B mixture (Lonza, 17--745E) containing 25 ng/mL macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN, USA, 416--ML-050) at 37 °C in 5% CO2 for 4‒5 days for differentiation. LPS (InvivoGen, San Diego, CA, USA, tlrl-eblps), zymosan (InvivoGen, tlrlzyn), ATP (Sigma‒Aldrich, A5394), nigericin (Sigma‒Aldrich, SML1779), AICAR (Sigma‒Aldrich, A9978), Baf-A1 (Sigma‒Aldrich, b1793), DBeQ (Selleckchem, Houston, TX, USA, S7199), SMER28 (Tocris Bioscience, Bristol, UK, 307538-42-7), and MitoTEMPO (Sigma‒Aldrich, SML0737) were added at the indicated concentrations and times for individual experiments.
Bulk RNA-seq and analysis of mouse-derived macrophagesTotal RNA from mouse BMDMs was extracted via TRIzol according to the manufacturer’s instructions. An Agilent TapeStation 4000 system (Agilent Technologies) was used to assess RNA quality, and an ND-2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) was used for RNA quantification. The library was constructed following the guidelines provided by the manufacturer and involved the use of the QuantSeq 3’ mRNA-Seq Library Prep Kit (Lexogen, Vienna, Austria, 225.96). Reverse transcription was performed using an oligo-dT primer that contained an Illumina-compatible sequence at the 5’ end, which hybridized to the RNA. Following the degradation of the RNA template, the initiation of second-strand synthesis occurred via a random primer with an Illumina-compatible linker sequence attached to its 5’ end. The double-stranded library was cleansed via magnetic beads. The library was amplified by incorporating the full adapter sequences necessary for cluster creation. The completed library was purified to remove PCR components. The high-throughput sequencing process was performed as follows: 75 single-end sequencing was performed at 10 M. The sequencing machine used was a NextSeq 550 (Illumina). The QuantSeq 3’ mRNA-Seq reads were aligned via Bowtie2, as described by Langmead and Salzberg in 2012. Bowtie2 mapped the reads to the reference genomes mm10 and UCSC, and the read counts were calculated via Bedtools (Quinlan AR, 2010). The read count data were normalized via the TMM + CPM normalization approach via EdgeR in R (R Development Core Team, Vienna, Austria) via Bioconductor [99]. The normalized counts were used for Z score calculation.
Quantitative real-time polymerase chain reaction (qRT‑PCR)qRT‒PCR was performed via SYBR Green Master Mix (Qiagen, 204074) in the Rotor-Gene Q 2plex system (Qiagen). The 2ΔΔ threshold cycle method was used for data analysis. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) was used for normalization. The sequences of primers used in this study are listed in Supplementary Table 4.
Enzyme-linked immunosorbent assay (ELISA)Cell supernatants from BMDMs were stored at -80 °C until ELISA. A mouse TNF-α ELISA kit (BD Biosciences, Franklin Lakes, NJ, USA, 558534), an IL-6 ELISA kit (BD Biosciences, 555240), an IL-1B ELISA kit (Invitrogen, 88-7013-88), and a human TNF-α ELISA kit (BD Biosciences, 555212) were used for the ELISA. Both the ELISA experiment and data analysis were conducted in accordance with the manufacturer’s protocols.
Western blottingThe cells were washed with cold phosphate-buffered saline (PBS) and lysed in Laemmli’s 5× Sample Buffer (ELPIS Biotech, EBA-1052) diluted with radioimmunoprecipitation assay (RIPA; LPS solution, Daejeon, South Korea, CRB002) buffer supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland, 11836153001) and phosphatase inhibitor cocktail (Roche, 4906837001) to obtain protein samples. Equal amounts of protein were boiled for 10 min on a heating block and cooled on ice for 10 min. Denatured protein samples were then subjected to sodium dodecyl sulfate‒polyacrylamide gel electrophoresis. The separated proteins were transferred to polyvinylidene difluoride (PVDF; Millipore, Billerica, MA, USA, IPVH0001) or nitrocellulose (NC; Pall Corporation, NY, USA, 66485) membranes. The membranes were incubated with primary antibodies at 4 °C overnight, followed by incubation with the corresponding horseradish peroxidase-conjugated secondary antibodies. The SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, 34095) was used for the visualization of appropriate signals in the iBright 750 imaging system (Thermo Fisher Scientific, CL750). The primary antibodies used were as follows: anti-phospho-NF-κB p65 (1:2000; Cell Signaling Technology, Danvers, MA, USA, 3033), anti-phospho-AKT (1:1000; Cell Signaling Technology, 9271), anti-phospho-JNK (1:0000; Cell Signaling Technology, 4668), anti-phospho-p44/42 MAPK ERK1/2 (1:2000; Cell Signaling Technology, 9101), anti-phospho-p38 (1:2000; Cell Signaling Technology, 4511), anti-beta-ACTB (1:2000; Santa Cruz Biotechnology, Dallas, TX, USA, sc-47778), anti-mature-IL1B (1:1000; Cell Signaling Technology, 12507), anti-mature-CASP1 (1:2000; Cell Signaling Technology, 2225), anti-NLRP3 (1:1000; Adipogen Life Sciences, San Diego, CA, USA, AG-20B-0014), anti-pro-IL1B (1:1000; Cell Signaling Technology, 12242), anti-pro-CASP1 (1:2000; Santa Cruz Biotechnology, sc-56036), anti-UBXN6 (1:1000; Abcam, ab103651), anti-CASP11 (1:1000; Cell signaling Technology, 14340), anti-LC3 (1:1000; Sigma‒Aldrich, L8918), anti-SEL1L (1:1000; Abcam, Cambridge, UK, ab78298), anti-SYVN1 (1:1000; Thermo Fisher Scientific, pa5-100081), anti-phospho-AMPK (1:1000; Cell Signaling Technology, 2535), anti-AMPK (1:1000; Cell Signaling Technology, 2532), anti-phospho-mTOR (1:1000; Cell Signaling Technology, 5536), anti-mTOR (1:1000; Cell Signaling Technology, 2983), anti-phospho-S6K1 (1:1000; Cell Signaling Technology, 9205), anti-S6K1 (1:1000; Cell Signaling Technology, 2708), and anti-LAMP1 (1:1000; Cell Signaling Technology, 99437). The secondary antibodies used were as follows: anti-rabbit IgG-horseradish peroxidase (HRP) linked (1:2000; Cell Signaling Technology, 7074), anti-mouse IgG-HRP linked (1:2000; Cell Signaling Technology, 7076), and anti-rat IgG-HRP linked (1:2000; Cell signaling Technology, 7077).
Immunofluorescence staining and confocal microscopic analysisThe cells were fixed in 4% paraformaldehyde (PFA) at room temperature (RT) overnight and permeabilized with 0.25% (v/v) Triton X-100 (Sigma‒Aldrich, T8787) in PBS for 10 min at RT. The cells were washed with PBS and then incubated with secondary antibodies for 2 h at RT. The primary antibodies used were as follows: anti-LC3 (1:400; MBL Co., LTD., Tokyo, Japan, PM036), anti-LAMP1 (1:400; Santa Cruz Biotechnology, sc-19992), anti-RELA/NF-κB (1:400; Santa Cruz Biotechnology, sc-8008), and anti-TFEB (1:400; Bethyl Laboratories, Montgomery, TX, USA, A303--673A). The secondary antibodies used were as follows: Alexa Fluor 488-conjugated anti-rabbit IgG (1:400; Invitrogen, A11006), Alexa Fluor 488-conjugated anti-rat IgG (1:400; Invitrogen, A11008), and Alexa Fluor 484-conjugated anti-mouse IgG (1:400; Invitrogen, A11029). For mitophagy analysis, the cells were stained with 100 nM MitoTracker Deep Red (Thermo Fisher Scientific, M22426) in prewarmed DMEM for 30 min at 37 °C, fixed in 4% PFA, permeabilized with 0.25% (v/v) Triton X-100, and then stained with anti-LC3 polyclonal antibodies. For the measurement of mtROS or cellular ROS, the cells were incubated with 1 μM MitoSOX Red (Thermo Fisher Scientific, M36008) or 20 μM DCF-DA (Calbiochem, Darmstadt, Germany, 287810) in DMEM for 20 min at 37 °C, fixed in 4% PFA, and permeabilized with 0.25% (v/v) Triton X-100. These cells were then mounted with Fluoromount-G, with DAPI (Invitrogen, 00-4595-52). For the immunostaining of in vivo paraffin sections, mouse lung tissues were harvested, fixed with 10% formalin, and embedded in paraffin wax. Paraffin sections (3 μm) were cut and immunostained with anti-Ly6G monoclonal antibodies (1:400; Bio X Cell, Lebanon, NH, USA, BE0075), anti-IL6 monoclonal antibodies (1:400; Santa Cruz Biotechnology, sc-57315), and appropriate secondary antibodies. ProLong™ Gold Antifade Mountant with DAPI (Invitrogen, P36931) was used for mounting. After 2 days of mounting, the images were visualized and captured via confocal microscopy (Carl Zeiss A.G., Baden-Württemberg, Germany, LSM 900 with Airyscan 2) and accompanying software (Zen blue edition; Carl Zeiss A.G.). The image capture parameters, such as excitation, emission, and exposure time, were kept constant. Each condition was assayed in quadruplicate, and at least 50–100 cells per field were counted. FIJI software was used to quantify LC3 puncta, fluorescence intensities, colocalization tests, and nuclear translocation levels in images via plugins, such as measurements, Pearson correlation coefficients, or EzColocalization.
Inflammasome analysisBMDMs were primed with LPS (100 ng/mL) in Opti-MEM (Gibco, 31985-070) for 4 h and then stimulated with 5 mM ATP (Sigma‒Aldrich, A5394) or 10 µM nigericin sodium salt (Sigma‒Aldrich, SML1779) for 45 min to activate the canonical inflammasome or transfected with 2 μg/mL LPS using Xfect polymer (Clontech Laboratories, Mountain View, CA, USA, 631318) according to the manufacturer’s instructions for the indicated times to activate the noncanonical inflammasome. The supernatants were collected and centrifuged at 4 °C to eliminate debris. Proteins were precipitated from the supernatant via StrataClean Resin (Agilent Technologies, 400724). Pull-down proteins were resuspended in 1× sample buffer diluted in RIPA buffer, boiled for 10 min, and subjected to Western blotting.
Transmission electron microscopy (TEM) analysisThe samples were sequentially fixed in 3% glutaraldehyde and 1% osmium tetroxide, cooled on ice for 1 h, washed with 0.1 M cacodylate buffer (pH 7.2) containing 0.1% CaCl2, and dehydrated in an ethanol and propylene oxide series. Next, the samples were embedded in the Epon 812 mixture and polymerized at 60 °C for 36 h. Using a ULTRACUT UC7 ultramicrotome (Leica Biosystems, Wetzlar, Germany), 70 nm thick sections were cut and mounted on 75-mesh copper grids. The sections were counterstained with uranyl acetate and lead citrate for 10 min and 7 min, respectively, and examined via KBSI Bio-High Voltage EM (JEM1400 Plus at 120 kV and JEM-1000BEF at 1000 kV; JEOL Ltd., Tokyo, Japan).
Production and transduction of lentiviral short hairpin RNA (shRNA)shRNA was produced via pLKO.1-based target shRNA plasmids. The plasmids pRSV-Rev (Addgene, Watertown, MA, USA, 12253), pMDLg/pRRE (Addgene, 12251), pMD2. G (Addgene, 12259), and UBXN6 or Foxo3 shRNA plasmids (Santa Cruz Biotechnology, sc-97428-SH or sc-37888-SH, respectively) were purchased for viral packaging. To produce the lentivirus, all the above plasmids were transfected into human embryonic kidney 293 T (HEK293T) cells via the Lipofectamine 2000 (Invitrogen, 11668-019) system for 72 h. Finally, the media supernatant containing the lentivirus was collected, centrifuged, and filtered before being stored at -80 °C. For lentiviral infection, BMDMs or human primary monocytes cultured in 48- or 96-well plates were infected with a lentiviral vector at a multiplicity of infection of 10 for 36 h, followed by subsequent treatment.
Untargeted metabolomics analysisIntracellular metabolic extracts were prepared from 2 × 106 cells with methanol containing internal standard solution (Human Metabolome Technologies, H3304-1002) and analyzed via capillary electrophoresis (CE)-connected electrospray ionization (ESI)-time-of-flight mass spectrometry (TOFMS) and a CE-tandem mass spectrometry (MS/MS) system (Human Metabolome Technologies, CARCINO-SCOPE). The culture medium was removed from the 60-mm dish, and the cells were washed twice in 5% mannitol solution (10 mL first and then 2 mL). The cells were then treated with 800 μL of methanol and 550 μL of Milli-Q water containing an internal standard solution. The metabolite extract was transferred into a microfuge tube and centrifuged at 2300 × g and 4 °C for 5 min. Next, the upper aqueous layer was centrifugally filtered through a Millipore 5-kDa cutoff filter at 9100 × g and 4 °C for 120 min to remove proteins. The filtrate was centrifugally concentrated and resuspended in 50 μL of Milli-Q water for CE-MS analysis. The concentrations of the metabolites were calculated by normalizing the peak area of each metabolite with respect to the area of the internal standard and by using standard curves, which were obtained via three-point calibrations.
Extracellular acidification rate (ECAR) analysisECAR measurements were performed via a Seahorse Bioscience XF24 Analyzer (Agilent Technologies). BMDMs were seeded at 2.5 × 105 cells per well in an XF24 cell culture microplate (Agilent Technologies, 100777-004), incubated overnight at 37 °C, and subsequently treated. Before analysis, 590 μL of assay medium (XF base medium containing 1 mM L-glutamine, 1 mM sodium pyruvate and 25 mM glucose [pH 7.4]) was added to each well, and the plate was incubated in a non-CO2 incubator for 1 h at 37 °C. The XF24 Biosensor Cartridge was activated for 24 h in XF calibrant solution (1 mL/well; Agilent Technologies, 100840--000) at 37 °C in a non-CO2 incubator. The basal ECAR was measured, and sequential injections of the following reagents were performed at 37 °C: the ATPase inhibitor oligomycin A (2 μg/mL; Sigma‒Aldrich, 75351), the uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP, 5 µM; Sigma‒Aldrich, C2759) and the mitochondrial complex I inhibitor rotenone (2 µM; Sigma‒Aldrich, 557368).
Measurement of branched-chain amino acids (BCAAs)BMDMs were seeded at 5 × 105 cells per well in 24-well plates (SPL Life Sciences, Pocheon, South Korea, 30024), incubated overnight at 37 °C, and stimulated with vehicle or LPS (100 ng/mL) for the indicated times. BCAAs were extracted via a BCAA assay kit (Abcam, ab83374) according to the manufacturer’s instructions, and the optical density (OD) at 450 nm was measured via a microplate reader (BMG Labtech, Ortenberg, Germany; LUMIstar Omega).
Experimental mouse models of sepsis and acute lung injury (ALI)To establish the LPS- or zymosan-induced sepsis model, the mice were intraperitoneally injected with LPS (Sigma‒Aldrich, L3755) at a dose of 14 or 20 mg/kg or with zymosan (InvivoGen, tlrlzyn) at a dose of 300 mg/kg. The mice were then observed for survival at 12 h intervals, and the overall survival rate was calculated until 5 days postinjection. To collect the tissues, the LPS-injected mice (14 mg/kg) were euthanized after 6 h. For the ALI model, the mice were anesthetized and intranasally administered LPS (10 mg/kg). Twenty-four hours after the injection, the lung tissues were collected and processed. To establish an immunosuppression model via a two-hit approach, the mice were subjected to CLP or a sham operation. After 24 h, the mice were intravenously injected with the P. aeruginosa reference strain PAO1 (3 × 106 CFU/head). At 4 h postinfection, blood, lung, and spleen tissues were collected to assess the bacterial burden and the levels of proinflammatory cytokines and chemokines. The remaining mice were then observed for survival at 12 h intervals, and the overall survival rate was calculated until 5 days post infection.
Histology and immunofluorescenceLung and spleen tissues were harvested and fixed in 10% formalin. After fixation, the tissues were embedded in paraffin wax. The paraffin-embedded tissues were cut into 4 μm thick sections. These sections were then stained with hematoxylin and eosin to visualize the tissue morphology. Images of the stained tissue sections were captured via light microscopy. Whole fields of tissue were scanned, and the inflamed areas in the lungs, as well as the ratio of red-to-white pulp in spleen tissues, were quantified. The percentage of inflamed area per whole field of tissue was determined via FIJI software.
Statistical analysisStatistical analysis was conducted via Prism 8.0 for Windows (GraphPad Software Inc., San Diego, CA, USA). Two-tailed Student’s t test was used to compare two groups, and one-way ANOVA with Tukey’s multiple comparison test or two-way ANOVA with Sidak’s multiple comparison test was used for three or more groups. The log-rank (Mantel‒Cox) test was used to determine the survival rate. The data are presented as the means ± standard deviations (SDs) or ± standard errors of the means (SEMs). Statistical significance is indicated as *p < 0.05, **p < 0.01, and ***p < 0.001.
留言 (0)