
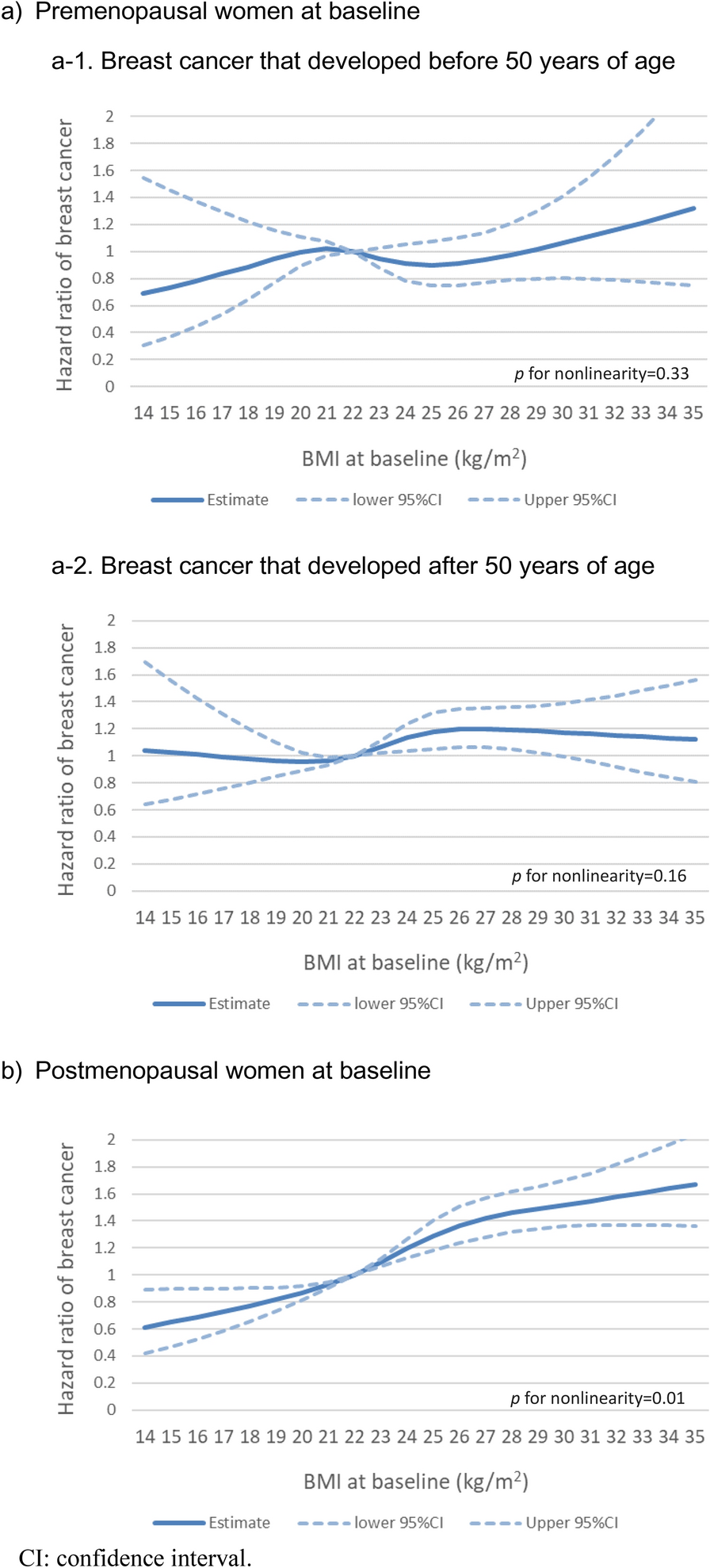
記住我







The anti-HER2 targeted therapy trastuzumab has been reported to induce autophagy in breast cancer cell lines in vitro [40]. To confirm that genetic downregulation of HER2 recapitulates the effects of pharmacological HER2 inhibition, tumor cells from a primary mammary adenocarcinoma in an MTB/TAN mouse [13] were cultured in the presence of doxycycline to maintain HER2 levels. Doxycycline withdrawal induced acute HER2 downregulation, which was accompanied by an acute 3.0-fold increase in levels of LC3-II, the cleaved, lipidated form of rat microtubule-associated protein 1 light chain 3 (LC3) that serves as a marker for autophagy (Fig. 1A and [41]). Consistent with the induction of autophagy, electron microscopy revealed an increased number of double-membraned autophagosomes in tumor cells following doxycycline withdrawal (Fig. 1, B and C).
Fig. 1
HER2 down-regulation induces autophagy in primary tumor cells in vitro. (A-E) MTB/TAN primary tumor cells subjected to doxycycline withdrawal for 24 h. Treatment with 50 µM chloroquine (CQ) for 24 h was used as a positive control for the induction of autophagy. (A) HER2 levels and conversion of LC3-I to LC3-II determined by western blotting. β-tubulin is shown as a loading control. (B) Representative images of doublemembraned autophagosomes (arrows) visualized by electron microscopy. Bar = 500 nm. (C) Quantification of autophagosomes per cell in (B). (D) Representative images of subcellular localization of EGFP-LC3 visualized by fluorescence microscopy. Original magnification, x400. (E) Quantification of average EGFP-LC3 punctae per cell in (D). Data represent mean ± SEM **P < 0.001, ***P < 0.0001
To confirm these results, we generated MTB/TAN primary tumor cells stably expressing the autophagy marker EGFP-LC3. Induction of autophagy induces the incorporation of cleaved, lipidated LC3-II into autophagosomes, which alters LC3 subcellular localization from diffusely cytoplasmic to punctate [41]. Doxycycline withdrawal from EGFP-LC3 expressing primary tumor cells resulted in an increase in the number of EGFP-positive punctae per cell compared to cells grown in the presence of HER2 (Fig. 1, D and E). Together, these results indicate that acute HER2 downregulation triggers autophagy in vitro.
To determine whether cytoplasmic contents sequestered in autophagosomes reached the lysosome and were degraded in primary MTB/TAN tumor cells, acute doxycycline withdrawal was combined with chloroquine treatment. Chloroquine (CQ) raises lysosomal pH and inhibits protein degradation within the autolysosome such that cells with flux through the autophagic pathway show additional increases in LC3-II levels when treated with chloroquine [34, 42]. Indeed, combined chloroquine treatment and HER2 downregulation in primary MTB/TAN tumor cells further augmented LC3-II levels beyond those in cells subjected to HER2 downregulation alone (Figs. 1A, 4.2-fold for combined chloroquine treatment and HER2 downregulation vs. 3.0-fold for HER2 downregulation alone). These findings suggest that acute HER2 downregulation induces autophagy as well as flux through the autophagic pathway.
We next asked whether acute HER2 downregulation induces autophagy in vivo. Doxycycline was withdrawn from MTB/TAN mice bearing primary mammary tumors for 48 h to downregulate HER2 expression and immunoblotting was used to analyze levels of p62/SQSTM1, which recognizes ubiquitin-marked proteins and sequesters them for degradation through autophagy. As a consequence, p62 itself is degraded in cells undergoing autophagy [43, 44]. Consistent with our in vitro observations, acute HER2 downregulation resulted in a rapid 59.9% reduction in p62 protein levels (Fig. 2A). Furthermore, primary orthotopic tumors generated from MTB/TAN tumor cells expressing EGFP-LC3 exhibited an increase in the number of EGFP-positive punctae per tumor cell following acute doxycycline withdrawal (Fig. 2B). These results indicate that autophagy is induced in primary tumor cells following acute HER2 downregulation.
Fig. 2
HER2 down-regulation induces autophagy in primary and dormant tumor cells in vivo. (A) HER2 and p62 levels evaluated by western blotting in orthotopic MTB/TAN primary tumors in the presence of doxycycline or following doxycycline withdrawal for 2 d. β-tubulin is shown as a loading control. (B) Representative images of subcellular localization of EGFP-LC3 in MTB/TAN primary orthotopic tumors or orthotopic tumors subjected to 2 d or 28 d of doxycycline withdrawal. Original magnification, x400
In MTB/TAN mice bearing primary mammary tumors, HER2 downregulation results in tumor regression to a non-palpable state, but leaves behind a small number of residual tumor cells that survive HER2 downregulation and persist in a dormant state in residual lesions within the mammary gland (Supplemental Fig. 1) [6, 14]. These cells are quiescent, yet remain competent to resume growth, resulting in recurrent tumors [6, 13, 14, 16]. Fluorescence microscopy performed on dormant EGFP-LC3-labeled tumor cells within residual lesions in mice harboring fully regressed orthotopic tumors revealed an increase in the number of EGFP-positive punctae per tumor cell compared to actively growing orthotopic primary tumors (Fig. 2B). Together, these observations suggest that autophagy is triggered in tumor cells following acute HER2 downregulation in vivo and in vitro and that dormant residual tumor cells undergo autophagy in vivo.
Pharmacological or genetic inhibition of autophagy inhibits mammary tumor recurrenceOur observations that autophagy occurs in vivo, is triggered by acute HER2 downregulation in actively growing primary tumor cells in mice, and persists in dormant mammary tumor cells were equally consistent with models in which autophagy is tumor suppressive or tumor promoting. To distinguish between these possibilities, we first treated MTB/TAN tumor-bearing mice with chloroquine. As chloroquine has been used safely in millions of people worldwide for the prevention and treatment of malaria and has a favorable therapeutic index, this drug represents an attractive approach to inhibiting autophagy in vivo [45].
To determine the effect of chloroquine treatment on mammary tumor recurrence, female nu/nu mice maintained on doxycycline were injected orthotopically with primary MTB/TAN tumor cells. Following primary tumor formation, tumor regression was induced by doxycycline withdrawal and HER2 downregulation [13]. Daily treatment with chloroquine was initiated to coincide with HER2 downregulation (Fig. 3A). Mice bearing fully regressed primary tumors were then monitored for recurrence. Daily chloroquine administration markedly delayed the onset of tumor recurrence in mice, with the median latency for tumor recurrence increasing from 84 to 140 days (H.R. = 3.12, 95% CI 1.45–6.72, P = 0.004; Fig. 3A). This finding suggests that autophagy promotes, rather than inhibits, mammary tumor recurrence.
Fig. 3
Pharmacological or genetic inhibition of autophagy inhibits mammary tumor recurrence. (A) Schematic of orthotopic recurrence model and timing of chloroquine treatment. Recurrence-free survival of female nu/nu mice harboring MTB/TAN orthotopic primary tumors induced to regress by doxycycline withdrawal and treated with vehicle (n = 24) or 60 mg/kg/d chloroquine (n = 26), as described. (B) Schematic and recurrence-free survival of female nu/nu mice harboring fully regressed orthotopic tumors derived from vector control (n = 14), shRNA targeting Atg5 (shAtg5; n = 9), or shRNA targeting Atg7 (shAtg7; n = 11)-expressing MTB/TAN tumor cells. Median recurrence latencies are indicated
Since pharmacological agents may have off-target effects, to confirm and extend these results we determined the effect on tumor recurrence of genetically inhibiting autophagy by knocking down the expression of ATG5 or ATG7, each of which is required for autophagy [46]. ATG5 and ATG7 are components of a ubiquitin-like conjugation system wherein the E1-like molecule ATG7 and the E2-like molecule ATG10 covalently link ATG5 to ATG12. ATG5-ATG12 then forms a complex with ATG16 that is required for formation of the autophagosome.
To determine if genetically inhibiting autophagy delays tumor recurrence, MTB/TAN primary tumor cells were generated that expressed shRNAs targeting either Atg5 or Atg7. qPCR and immunoblotting confirmed knockdown of ATG5 and ATG7 (Supplemental Fig. 2). These cells were then injected orthotopically into the mammary glands of nu/nu mice on doxycycline, as were cells transduced with a vector control, to form primary tumors. As above, doxycycline was withdrawn from mice bearing orthotopic tumors of the same size to induce HER2 downregulation, which resulted in the regression of tumors to a non-palpable state. Mice were then monitored for recurrence.
This analysis revealed that genetic inhibition of autophagy by knocking down either ATG5 or ATG7 dramatically impaired tumor recurrence (H.R. = 5.35, 95% CI 1.72–16.62, P = 0.004; H.R.= 7.56, 95% CI 2.33–24.58, P < 0.001; Fig. 3B). These results are consistent with the effects of autophagy inhibition by chloroquine and further suggest that autophagy is required for the recurrence of HER2-induced tumors.
Beclin 1 is required for mammary tumor recurrenceThe observations that ATG5 knockdown, ATG7 knockdown, and chloroquine treatment each result in a delay in mammary tumor recurrence is consistent with a model in which autophagy plays a pro-tumorigenic role in breast cancer recurrence. However, multiple lines of evidence suggest that autophagy is tumor suppressive, several of which are derived from studies in which Becn1 was monoallelically deleted in tumor cells or in mice [26, 28, 30, 31, 47]. BECLIN1 is a haploinsufficient tumor suppressor whose heterozygous loss in mice results in increased susceptibility to lymphomas, liver cancer, lung cancer, and mammary epithelial hyperplasias [28, 47]. BECLIN1 is also essential for the initiation of autophagosome formation by means of its ability to form a complex with the Vps34 PI3 kinase and other proteins [20]. Since knockdown or loss of BECLIN1 inhibits autophagy while increasing susceptibility to primary tumor formation, these data suggest a tumor suppressive role for BECLIN1 and autophagy.
In light of these conflicting data, we wished to determine whether – consistent with a tumor suppressive role for BECLIN1 and autophagy – loss of BECLIN1 would accelerate mammary tumor recurrence or whether – consistent with a tumor promoting role for autophagy as suggested by the effects of chloroquine treatment as well as ATG5 or ATG7 knockdown – loss of BECLIN1 would inhibit mammary tumor recurrence. To address this question, Becn1+/- mice were crossed to MTB/TAN inducible bitransgenic mice to generate cohorts of MTB/TAN; Becn1+/+ and MTB/TAN; Becn1+/- female mice. Primary mammary tumors were then induced by chronic activation of HER2 via doxycycline administration.
The incidence, latency, multiplicity and growth rate of primary HER2-induced tumors did not differ between Becn1+/+ and Becn1+/- mice (Fig. 4A and data not shown). Tumor-bearing mice were then deinduced and mice bearing fully regressed tumors were monitored for recurrence. The rate of primary tumor regression was unaffected by Becn1 genotype (data not shown). In contrast, deletion of one allele of Becn1 markedly delayed tumor recurrence with the median latency for tumor recurrence increasing from 77 days to 131 days (H.R. = 2.38, 95% CI 1.15–4.94, P = 0.019; Fig. 4B). These results demonstrate that BECLIN1 is required for mammary tumor recurrence and further support our findings based on ATG5 or ATG7 knockdown and chloroquine treatment that autophagy is required for mammary recurrence.
Fig. 4
Beclin 1 is required for mammary tumor recurrence in intact mice. (A) Primary tumor-free survival of female MTB/TAN; Beclin 1+/+(n = 38) and MTB/TAN; Beclin 1+/- mice (n = 40) administered doxycycline to induce HER2 beginning at 6 wk of age. (B) Recurrence-free survival of female MTB/TAN; Beclin 1+/+(n = 18) and MTB/TAN; Beclin 1+/- mice (n = 18) that harbored fully regressed primary tumors following doxycycline withdrawal. Median recurrence latencies are indicated
Selection against dormant tumor cells with impaired autophagyHaving established a role for autophagy in the recurrence of HER2-induced mammary tumors, we wished to determine the cellular basis for this requirement. Based on evidence supporting a role for autophagy in responses to cellular stress, we considered the possibility that autophagy contributes to the survival of tumor cells subjected to HER2 pathway inhibition. To test this hypothesis, HER2 was downregulated for 24 h in vitro in MTB/TAN primary tumor cells expressing an shRNA targeting Atg5 or a control vector and the percentage of viable cells was determined. The survival of tumor cells transduced with a control vector was not altered by HER2 downregulation in the presence of 0.5% serum (Fig. 5A). In contrast, a substantial increase in cell death was observed in tumor cells expressing HER2 when ATG5 was knocked down, and the combination of ATG5 knockdown with HER2 downregulation resulted in a dramatic impairment in cell survival due to increased apoptosis (Fig. 5A and data not shown). These findings suggest that autophagy is required for the survival of primary mammary tumor cells subjected to acute HER2 downregulation in vitro, which is consistent with a prior report [40].
Fig. 5
Atg5 is required for dormant mammary tumor cell survival. (A) Viability of primary MTB/TAN tumor cells expressing an shRNA targeting Atg5 (shAtg5) or empty vector grown in 0.5% serum with or without doxycycline for 24 h. Data represent mean ± SEM *P < 0.05. (B-D) Mammary fat pads of female nu/nu mice on doxycycline were injected with an equal ratio of primary MTB/TAN tumor cells that were either transduced with shAtg5 and labeled with H2B-mCherry or were transduced with a vector control and labeled with H2B-EGFP. (B) Schematic of orthotopic competition assay and timing of tumor harvest. (C) Representative fluorescence microscopy images of primary tumors (n = 6), residual lesions 14 d post-deinduction (n = 5), residual lesions 28 d post-deinduction (n = 5), and recurrent tumors (n = 3) from (B). Magnification, x400. (D) Percentage of H2B-EGFP-positive and H2B-mCherry-positive tumor cells was determined at each time point in (B). Data represent mean ± SEM ***P < 0.0001
We next wished to test the role of autophagy in promoting tumor cell survival in vivo during the process of mammary tumor recurrence. MTB/TAN primary tumor cells expressing an shRNA targeting Atg5 were labeled with an H2B-mCherry reporter, whereas MTB/TAN primary tumor cells transduced with an empty vector were labeled with an H2B-EGFP reporter. These two fluorescent populations of isogenic cells were admixed in equal parts (Supplemental Fig. 3), injected into the mammary glands of nu/nu mice maintained on doxycycline, and allowed to generate orthotopic primary tumors. Doxycycline was then withdrawn to initiate the process of tumor regression and fluorescence microscopy was used to determine the ratio of mCherry-labeled ATG5-knockdown cells to EGFP-labeled control cells in primary tumors, residual tumor lesions 14 days or 28 days following HER2 downregulation, and recurrent tumors (Fig. 5, B-D).
As predicted based on the unperturbed nature of HER2-induced primary tumorigenesis in Becn1+/- mice, we observed no selection for or against cells with ATG5 knockdown in primary tumors (Fig. 5, C and D). Furthermore, we observed no selection for or against cells expressing an Atg5 shRNA in residual lesions 14 days post-HER2 downregulation, a time point that roughly corresponds to the completion of tumor regression and early stages of tumor dormancy (Fig. 5, C and D and Supplemental Fig. 1). In contrast, cells with ATG5 knockdown were present at a lower than expected ratio in residual lesions 28 days following HER2 downregulation, a time point at which residual tumor cells have been in a dormant state for ∼ 3 wks (Fig. 5, C and D and Supplemental Fig. 1). This competitive disadvantage was even more pronounced in recurrent tumors, in which only 1% of fluorescent cells were mCherry-positive (Fig. 5, C and D).
To rule out differential effects of the H2B-mCherry and H2B-EGFP reporters, this experiment was repeated after exchanging fluorescent protein labels between Atg5 shRNA and control cells. This confirmed the pronounced competitive disadvantage of Atg5 shRNA tumor cells compared with control cells during late dormancy and recurrence time points (Supplemental Fig. 3). Together, these data indicate that primary tumor cells in which autophagy is impaired are at a strong, cell-intrinsic selective disadvantage following HER2 downregulation, but not until tumor cells have entered a dormant state.
Autophagy is required for dormant residual tumor cell survival in vivoOur observations to this point suggested that autophagy is required for the survival of dormant tumor cells following oncogene downregulation. To test directly whether tumor dormancy represents a cellular state in which cells are particularly reliant upon autophagy for their survival, we determined the impact of chloroquine treatment on dormant tumor cells generated by HER2 downregulation. Primary MTB/TAN tumor cells were grown in 10% serum plus doxycycline, 1% serum plus doxycycline, or 1% serum in the absence of doxycycline. After 3 wk, cells grown in 1% serum in the absence of doxycycline were Ki67-negative, but could be induced to reenter the cell cycle solely by the re-addition of doxycycline, demonstrating that these cells are reversibly growth arrested (Supplemental Fig. 4).
After 3 wk in the above media, cells were treated with chloroquine for 1 week and cell viability was assessed. Dormant tumor cells maintained in 1% serum in the absence of HER2 expression were markedly more sensitive to chloroquine treatment than proliferating cells expressing HER2 maintained in either 1% serum or 10% serum (Fig. 6A). These findings suggest that dormant tumor cells are more dependent upon autophagy for survival than actively proliferating cells.
Fig. 6
Chloroquine delays recurrence by decreasing survival of dormant mammary tumor cells. (A) Viability of primary MTB/TAN tumor cells grown in 10% serum plus doxycycline, 1% serum plus doxycycline, or 1% serum without doxycycline for 3 wk, then treated with 50 µM chloroquine (CQ) or vehicle control for 1 wk. (B) Viability of MTB/TAN tumor cells grown in 1% serum without doxycycline for 3 wk, then treated with 50 µM chloroquine or 50 µM chloroquine plus 10 mM methylpyruvate (MP) for 1 wk. (C) Schematic of orthotopic dormancy model and timing of chloroquine treatment. Female nu/nu mice harboring primary orthotopic tumors generated from primary MTB/TAN cells were deinduced for 21 days and then treated daily with chloroquine 60 mg/kg (n = 21) or vehicle control (n = 23) for 2 wk. At 35 d post-deinduction, the number of EGFP-positive tumor cells per gland was determined using flow cytometry. Data represent mean ± SEM. **P < 0.001, *P < 0.05
We then tested if the degradation of metabolic substrates by autophagy was required to maintain oxidative phosphorylation and the viability of dormant mammary tumor cells treated with chloroquine. A cell-permeable form of pyruvate, methylpyruvate, was added to the culture media of dormant MTB/TAN tumor cells at the time of chloroquine treatment. Methylpyruvate, once taken up by a cell, can be oxidized in the tricarboxylic acid (TCA) cycle to fuel ATP production. The addition of methylpyruvate to dormant tumor cells rescued the cell death observed in response to chloroquine treatment (Fig. 6B). This suggests that the inhibition of autophagy decreased dormant tumor cell viability, at least in part due to the suppression of cellular bioenergetics.
We next wished to determine if dormant mammary tumors cells are dependent upon autophagy for survival in vivo. To address this, mice harboring orthotopic EGFP-labeled dormant residual tumor cells were treated with chloroquine for 2 wk beginning 21 d after HER2 downregulation. Mammary glands were then harvested, digested to form a single-cell suspension, and the number of EGFP-positive residual tumor cells was determined by flow cytometry. This analysis revealed that mice treated with chloroquine for 2 wk within the period of dormancy harbored 38% fewer dormant tumor cells (P < 0.05) than mice treated with a vehicle control (Fig. 6C). Given the absence of tumor cell proliferation at these time points, these data strongly suggest that autophagy is required for the survival of dormant mammary tumor cells in vivo.
Autophagy inhibition in mice bearing dormant minimal residual disease suppresses recurrenceOur findings to this point suggested that dormant residual tumor cells may be uniquely dependent upon autophagy for their survival. If correct, our data would predict that inhibitors of autophagy would be most effective not in actively growing tumors, but in dormant residual tumor cells. If so, chloroquine could be used on that basis to reduce the pool of viable dormant residual tumor cells and thereby prevent or delay tumor recurrence.
To test this hypothesis, we again employed chloroquine in an orthotopic recurrence assay, with the exception that chloroquine treatment was initiated 28 days following doxycycline withdrawal when tumors had completed their regression and residual tumor cells exist in a dormant state [6, 14], rather than at the time of HER2 downregulation when tumor cells are still proliferating and tumor regression has not yet begun (Fig. 7A). Mice were then monitored for tumor recurrence.
Fig. 7
Chloroquine treatment of mice bearing dormant minimal residual disease impairs recurrence. (A) Schematic of orthotopic recurrence model and timing of chloroquine treatment. (B) Recurrence-free survival of mice bearing fully regressed orthotopic tumors that were treated daily with vehicle control (n = 22) or 60 mg/kg chloroquine (n = 24) as described. Median latencies for tumor recurrence are indicated
This analysis revealed that autophagy inhibition restricted specifically to the period of tumor dormancy markedly delayed recurrence (H.R. = 2.88, 95% CI 1.39–5.98, p = 0.005; Fig. 7B). Notably, the magnitude of the effect of chloroquine administration beginning 28 days after HER2 downregulation was nearly identical to that observed for the effect of chloroquine administration beginning immediately at the time of HER2 downregulation (Fig. 3A). This finding was consistent with results from the fluorescent cell competition assay (Fig. 5, B-D and Supplemental Fig. 3), indicating that cells with impaired autophagy are not selected against during primary tumor formation or within the first 14 d following HER2 downregulation. Similarly, the magnitude of the effect of chloroquine administration beginning 28 days after HER2 downregulation was similar to that observed for the effect of deleting one allele of Becn1, which also failed to affect the rate of primary tumorigenesis in MTB/TAN mice (Fig. 4). Together, these observations provide further support for a model in which the effects of autophagy inhibition on tumor cell survival are largely confined to tumor dormancy.
留言 (0)