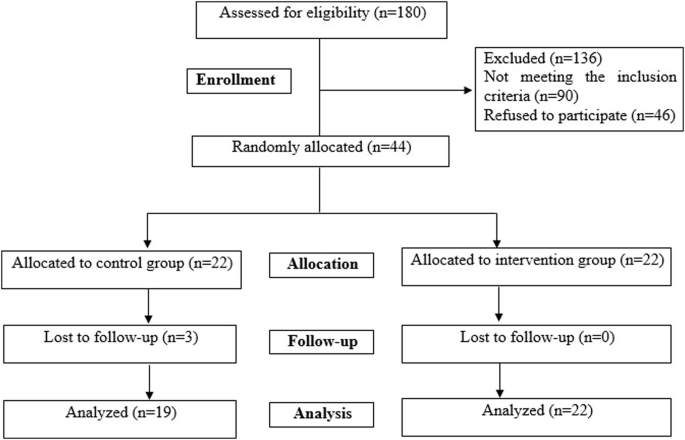
A diagram of the data collection procedures is shown in Fig. S1.
Animals
All animal experiments were approved by the Institutional Animal Care and Use Committee of ShanghaiTech University and were consistent with the governmental regulations of China for the care and use of animals.
GLP-1R knockout mice (GLP-1R KO, Shanghai Model Organisms, based on C57BL/6N) were generated using CRISPR/Cas9 technology, which causes Glp1r gene protein reading frame shift and loss of function by using non-homologous recombination repair to introduce mutations. Briefly, the process is as follows: Cas9 mRNA and gRNA were obtained by in vitro transcription; Cas9 mRNA and gRNA were microinjected into the fertilized eggs of WT mice to obtain F0 generation mice. 11 positive F1 generation mice were obtained by mating the F0 generation mice identified as positive by PCR amplification and sequencing with C57BL/6N mice. Mutant mice were genotyped to ensure the deletion of the target Glp1r segment.
Male WT mice and male GLP-1R knockout mice, aged 7–13 weeks with weighing 20–30 g were included in this study [29]. Animals were housed under a 12 h light/dark cycle at a room temperature of 22 ± 1 °C with 45% humidity, given ad libitum access to food and water.
Based on existing literature, an N of 6 was initially tested for DMRS in both groups and the fMRI group of GLP-1R KO mice, an N of 7 for PET in both groups [30,31,32,33], and an N of 6 for both groups for cyclic adenosine monophosphate (cAMP) measurement. For the fMRI group of WT mice, we used data from 11 WT mice recorded with an identical protocol from a previous, unpublished study. As detailed in later sections, the DMRS spectra of one WT mouse was excluded due to failure in data processing, the fMRI data from one GLP-1R KO mouse was excluded due to being an outlier, and one GLP-1R KO mouse died during PET image acquisition. Therefore, the total number of mice used herein is 52. Five WT mice and six GLP-1R KO mice were imaged with DMRS. Seven WT mice and six GLP-1R KO mice were imaged with [18F]-FDG-PET. Eleven WT mice and Five GLP-1R KO mice were imaged with rs-fMRI. Six WT mice and six GLP-1R KO mice had cAMP measured. As results were already strongly statistically significant with these initial values, we did not add additional mice to the study under the principle of reduction in animal experiments [34].
Animals were not used for multiple experiments as two of the three experiments were non-survival (due to the blood collection following DMRS and the urethane anesthesia used for fMRI) and incompatible (DMRS and PET both required labeled glucose injection, the DMRS and fMRI signals may interfere with each other, and fMRI required different anesthesia than the other two experiments).
Deuterium magnetic resonance spectroscopy
WT and GLP-1R KO mice were anesthetized by inhaling a mixture of 1–3% isoflurane and compressed air (1.5 L/min). The animals were not fasted prior to scanning. A 30 gauge needle and 70-cm-long venous catheter preflushed with a heparin-solution (50 IU/ml) solution was inserted into a lateral tail vein and fixed with adhesive tape to prevent blood clotting. During the entire experimental procedure, the animals were maintained under normal physiological conditions. Rectal temperature was maintained at 37 ± 1 °C using heated circulating water and a hot-air blower, and respiratory rate was monitored at ~80–120 breaths per minute.
DMRS experiments were performed in a 9.4 Tesla (BioSpec 90/30 USR; Bruker Biospin MRI, Ettlingen, Germany), using a custom-built 1H/2H birdcage radiofrequency (RF) coil (Larmor Device, Hangzhou, China) [35] as receiver and transmitter. The magnet was equipped with B-GA12SHP for BC70/20 gradient coil, and B-GA12SHP INSERT for BGA10SHP BC94/30 shimming coil. The 1H loop was tuned to 400.3 MHz for localizing the mouse brain and shimming, while the 2H loop was tuned to 61.4 MHz for acquiring 2H spectra. A deuterium water phantom (physiological saline solution containing 10% D2O) was used to calibrate the reference power at 60 W. In animal experiments, the coil center was positioned over the mouse brain. The 1_localizer sequence and 1H signal were employed for brain localization, and the 2_localizer_shim sequence was utilized for field homogenization. 2H-NMR spectra were obtained with the 2H loop and a single-pulse sequence, using saturation bands to suppress signals from tissues other than the brain. A single-pulse sequence was applied to acquire dynamic DMRS spectra from the mouse brain with the following parameters: 2 KHz bandwidth, 512 data points for the free induction decay (FID), 64° flip angle, 350 ms repetition time (TR) with 800 averages (5 min per spectrum) and a total of 15 spectra. For each mouse, baseline 2H spectra were obtained followed by around 1 min intravenous infusion of [6,6’]-2H2 glucose (d66, Aladdin; 1.95 g/kg, 1 M/L) and ~75 min continuous DMRS scanning.
For the first three mice, immediately following DMRS data collection, we took blood from the hearts to perform in vitro 2H-NMR with an 18.8 T nuclear magnetic resonance facility (800 MHz NMR) to validate our results recorded at a lower spectral resolution.
Mice were excluded if no peaks other than water could be distinguished from their spectrum. This resulted in the exclusion of one WT mouse’s data.
GLX and Lactate peaks were only visible in spectra for one WT mouse. An example of a spectrum from a single mouse with all four peaks visible is shown in Fig. S2. Therefore, our analysis focused on HDO and deuterium glucose. 2H resonance signal integrals (HDO and deuterium glucose) were fitted using MestReNova software (version 14.2.0, MestreLab Research, Spain) and MATLAB code for quantification. The 2H-NMR spectra were generated by 10 Hz exponential window function transformation, followed by zero-filling to generate 1024 FID data points (twice the original). Manual phase correction and multi-point baseline correction were performed, and the relevant steps were saved for batch processing. For 2H-NMR spectra, using deuterated water (HDO) as the chemical shift reference (~4.7 ppm), the peaks corresponding to d-glucose-6,6’-d2 were marked (around 3.8 ppm). These two signal peaks were fitted, and the area under the curve was extracted. According to known relaxation times from previous studies [23, 32], relaxation-corrected glucose concentrations were measured in dynamic DMRS. The decay equation for glucose is given in Eq. 1.
$$f\left(t\right)=Y0+\left(Y1-Y0\right)* ^}$$
(1)
In Eq. 1, \(f\) represents concentration, \(t\) represents time, \(e\) is the base of natural logarithms, \(Y0\) is the initial concentration of post-injection, \(Y1\) is the final concentration, and \(k\) is the rate constant, expressed in inverse minutes. The rate constant of brain glucose metabolism for each mouse was determined, and statistical analysis was performed on the two groups of samples using a Student’s t-test.
Positron emission tomography
Mice were fasted for 12 h before imaging. 2-deoxy-2-[fluorine-18] fluoro-d-glucose ([18 F]-FDG) (Dongcheng AMS Pharmaceutical atom, Shanghai, China) was injected into the tail vein at a dose of ~3.7 MBq/10 g body weight (0.1 mCi/10 g). [18F]-FDG was distributed in the mice for 40 min while awake. Mice were imaged using a Bruker BioSpec 9.4 T scanner with PET insert (Bruker, Ettingen, Germany) for [18F]-FDG-PET imaging. To prevent movement contamination from confounding the result with 18F-FDG uptake in muscles, isoflurane anesthesia (5% for induction and ≤2% for maintenance) was used here for biodistribution detection and imaging. First, a T2-weighted MRI (T2WI) rapid acquisition with relaxation enhancement (RARE) (echo time (TE) = 54.39 ms, repetition time (TR) = 1800 ms, field of view (FOV) = 35 × 35 × 85 mm3, matrix size = 128 × 128) was performed. Then, the mice were imaged with 10-min static PET scanning. PET images were reconstructed with the ordered-subsets expectation maximization 2D algorithm (OSEM2D) in Paravision 360. PMOD v4.4 Software (Bruker, Ettingen, Germany) was used for image registration and quantitative analysis. Standardized uptake value (SUV) was calculated [SUV = radioactivity activity concentration (KBq/ml)/body weight (g)/injected dose (kBq)] for semi-quantitative analysis. Afterward, SUV values were corrected for body weight [SUVbw = SUV × body weight (g/ml)].
Resting-state functional MRI (rs-fMRI)
Functional MRI data were recorded using a Bruker BioSpec 9.4 T scanner, equipped with an 86-mm volume coil for transmitting, and a 4-channel phased array cryogenic mouse head coil (Bruker, Ettingen, Germany) for receiving. Animals were anesthetized with 25% urethane dissolved in distilled water (Sigma, U2500–100 G) using intraperitoneal bolus injections (1.3 g/kg) divided into three separate doses. The interval time was 10 min between each dose [36,37,38]. No ventilation is needed to maintain the hemodynamic state of animals under urethane. Sufficient anesthesia was judged by no toe-pinch reflex or weak toe-pinch response. A built-in warm water heating pad (Bruker) was used to maintain body temperature between 36 and 37.5 °C. Respiratory rates were monitored throughout, and respiration was maintained within the range of 180–220 breaths per minute.
Anatomical images were acquired using a T2-Turbo-RARE scan, TR = 2230 ms, TE = 39.43 ms, FOV = 20 × 7.9 mm2, slices: 23, slice thickness = 0.3 mm, matrix size = 200 × 79. A gradient-echo EPI sequence, TR = 1000 ms, TE = 13 ms, FOV = 20 × 7.9 mm2, slices: 13, slice thickness = 0.6 mm, matrix size = 121 × 48, repetitions = 480, was used to obtain resting state functional MRI (rs-fMRI) blood oxygen level-dependent (BOLD) data.
rs-fMRI BOLD data were preprocessed and analyzed using custom-written scripts in MATLAB. Motion correction and slice timing were performed in SPM12 (Wellcome Trust Centre for Neuroimaging, London, UK). Anatomical images were registered to the mouse brain template [39], using a nonlinear registration (50 iterations, normalized mutual information, and otherwise default) with BioImage Suite (Yale School of Medicine, 2015; bioimagesuite.yale.edu). Each mouse’s corresponding nonlinear registration was then applied to rs-fMRI data from the same mouse. To facilitate inter-mice statistics, each rs-fMRI image was spatially smoothed (0.5 mm isotropic Gaussian kernel), and each voxel’s time series was bandpass filtered from 0.01 to 0.3 Hz [40, 41].
Per-voxel rs-fMRI metrics
The global signal is the average of the BOLD signal from all voxels, at each time point. Correlating the global signal with the time series of each voxel (Pearson’s correlation) yields a whole-brain correlation, which is also considered a “weighted” form of functional connectivity density [42]. A normalizing version of Fisher’s transformation was applied to normalize the Pearson correlation to a hypothetical N (0,1) distribution of Z scores [43]. Results were averaged together for all voxels within the whole brain.
The amplitude of low-frequency fluctuations (ALFF) in fMRI is a widely used metric for assessing spontaneous neuronal activity [44]. For each voxel, the power spectrum was computed from the fMRI time series, and then the square root of the power spectrum was obtained. The ALFF value was derived by averaging the square root values within the frequency range of 0.01–0.1 Hz [45]. Results were averaged together for all voxels within the whole brain.
Functional connectivity density (FCD) mapping, a metric derived from graph theory and employing rs-fMRI data, utilizes Pearson’s correlation to generate a map of functional connections in the brain [46]. Global functional connectivity density (gFCD) measures connections across the entire brain. For each voxel, we used a correlation threshold of r ≥ 0.25 to determine the number of voxels across the entire brain linked to it. The resulting gFCD values were calculated on a per-mouse basis. Results are averaged together for all voxels within the whole brain.
The global signal, ALFF, and FCD were tested in GraphPad (version 9.5.1 GraphPad Software, LLC) using the built-in ROUT method to find outliers. One GLP-1R KO mouse had outliers in all three metrics, and was thus excluded.
cAMP measurement
cAMP levels in whole brain tissues were measured using a cAMP assay kit (Shanghai Boke Biotechnology Co., Ltd., Shanghai, China). Each mouse was euthanized, and their entire brain was removed and liquified to use as a single sample. Data from all mice (N = 6 per group) were used. The assay was performed according to the manufacturer’s instructions provided in the kit.
Statistical analysis
We analyzed the distribution of these data and applied the F-test to test if the variances of the two populations were equal. Statistical analysis of the data was performed using a Student’s t-test. Statistical significance was set at p < 0.05. Effect size was used to quantify the differences between group means and the relationships between variables [47, 48]. The effect size was calculated here using the method of Cohen’s d for a t-test [49]. For non-normal distribution data, instead of standard deviation, we used the 84.13% percentile minus the median when we calculated the effect size [49]. Normally, from this calculation, effect sizes greater than 0.5 are considered a “large difference effect”. Detailed information regarding the testing for a normal distribution, F-test and statistical analysis results, and effect size results are summarized in Table S1.





留言 (0)