
記住我
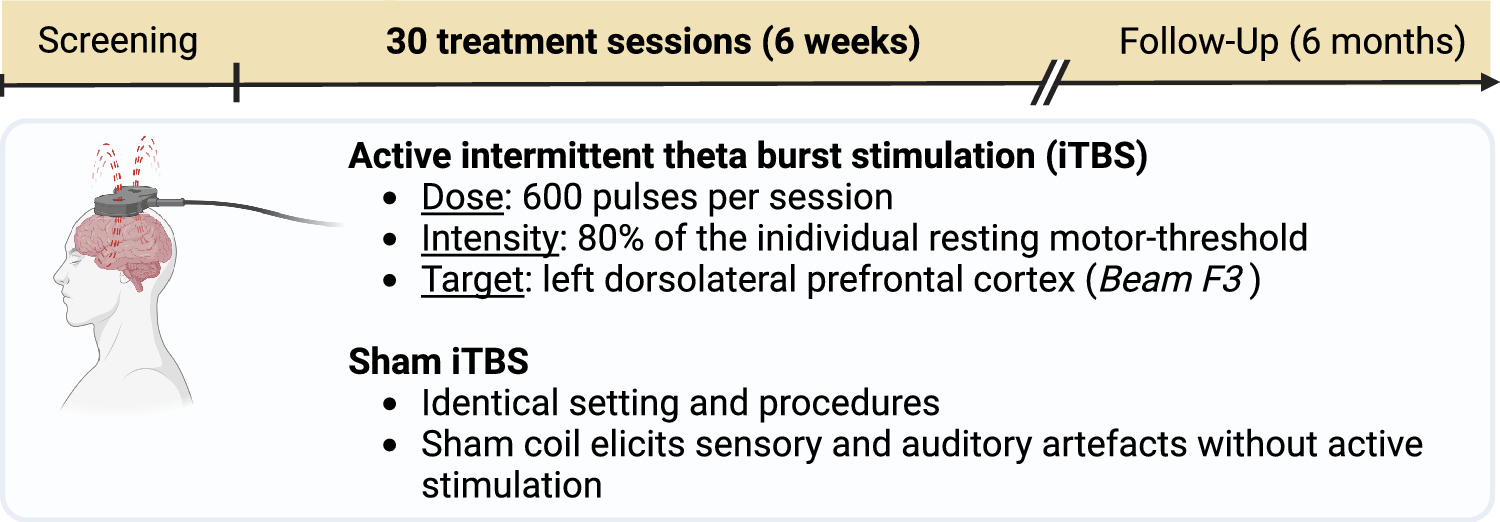

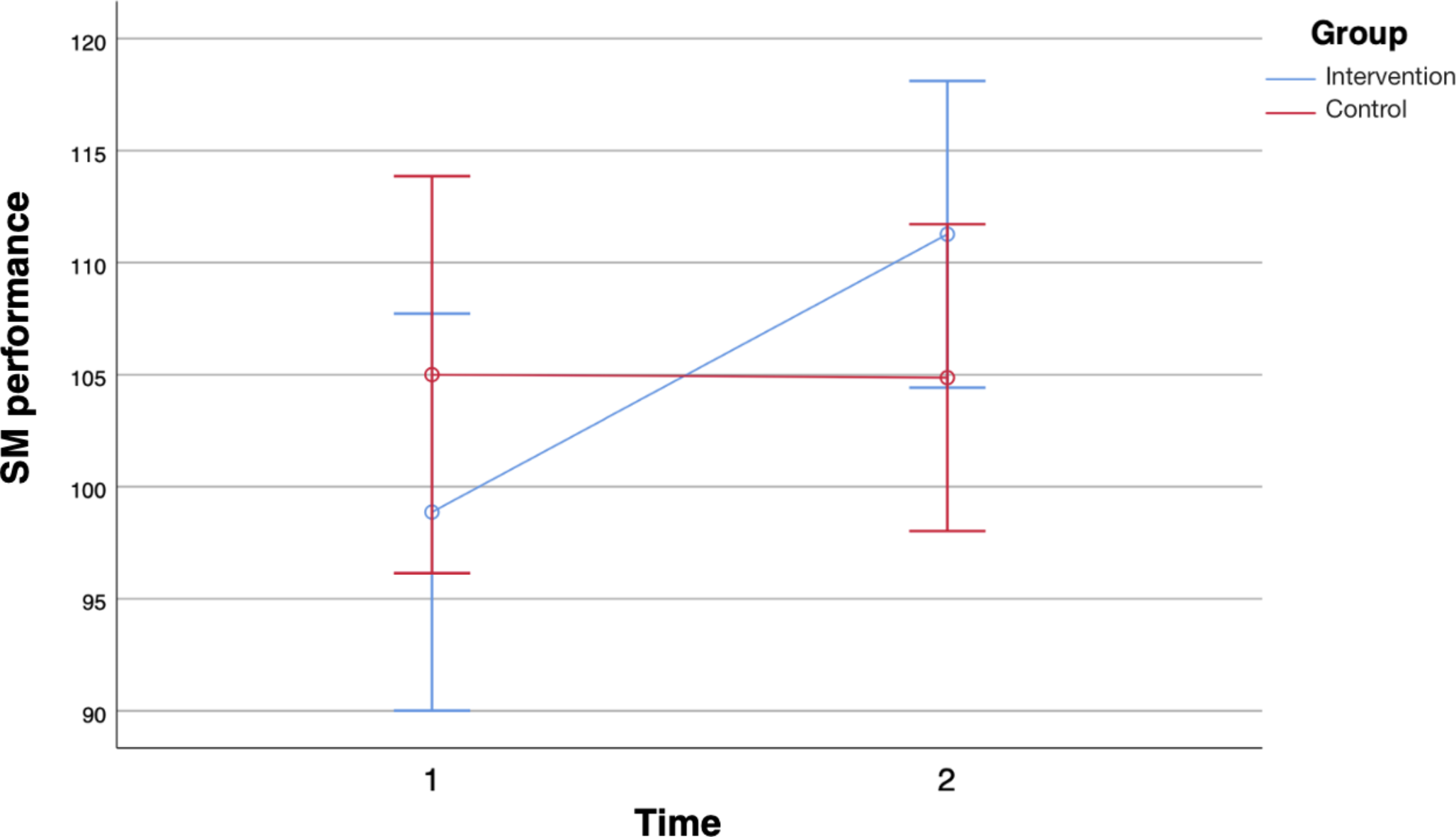


Table 1 presents an overview of measures that are assessed in this trial.
Table 1 Schedule of visitsPrimary outcomeThe primary outcome will be the difference in MADRS scores at week 6 (post-treatment visit) between active iTBS and sham iTBS, controlling for baseline MADRS scores. The MADRS is a clinician-rated measure of depression severity that assesses ten common depressive symptoms (apparent sadness, reported sadness, inner tension, reduced sleep, reduced appetite, concentration difficulties, lassitude, inability to feel, pessimistic thoughts, and suicidal thoughts) on a scale from 0–6. The overall score ranges from 0 to 60, with higher MADRS scores indicating more severe depression. We will use the structured interview guide for the MADRS (SIGMA)[30] to improve the reliability of assessments between study raters. All raters will receive video-based SIGMA training, during which they will rate two example video interviews.
Secondary, exploratory outcomesSecondary outcomes will explore group differences in (1) MADRS score changes from baseline to 3 and 6 months; (2) response rates (≥ 50% MADRS score reduction) at week 6, and 3 and 6 months; (3) remission rates (MADRS score ≤ 10) at week 6, and 3 and 6 months; (4) changes in self-reported depression, anxiety, and stress using the Depression, Anxiety, and Stress Scale, 21-item version (DASS-21)[31] at week 6, and 3 and 6 months; (5) the change of self-reported anhedonia using the Dimensional Anhedonia Rating Scale (DARS) [32] at week 6, and 3 and 6 months; and (6) change of self-reported health-related functioning using the World Health Organisation Disability Assessment Schedule (WHODAS 2.0) [33] at week 6, and 3 and 6 months.
Safety and tolerability outcomesSafety outcomes will be the group differences in (1) rates of reported adverse events (AEs); (2) change of vital signs (blood pressure and heart rate) and Body Mass Index (BMI); and (3) rates of reported instances of suicidality; all measured at week 6 (post-treatment visit). Suicidality will be assessed at every visit using the Columbia-Suicide Severity Rating Scale (C-SSRS) [34], an interviewer-rated scale that measures the current intensity of patients’ specific suicidal ideation on the day of assessment. A patient will be identified as experiencing acute suicidality if they affirm item 4, item 5, or both. Upon such identification, treatment will be halted immediately, and the patient will be directed to suitable crisis intervention services for urgent care. Tolerability outcomes will be the group differences in (1) rates of participants who discontinue the treatment; and (2) rates of participants who discontinue the treatment due to an AE.
Other assessmentsThe following variables and measures will be collected at baseline and/or during the course of the trial as potential moderators and mediators of treatment: Demographic and clinical variables (e.g., age and gender, prior psychiatric treatment, medical co-morbidities); self-reported symptoms of insomnia using the Insomnia severity index (ISI) [35]; self-reported experiences of adverse childhood experiences using the Childhood Trauma Questionnaire (CTQ) [36]; self-reported maladaptive personality traits, using the PID5BF + M, a validated, expanded German version of the Personality Inventory for DSM-5 (PID-5) [37]; self-reported participation in social relationships using the Social Network Index (SNI) [38]; self-reported loneliness using the UCLA Loneliness Scale (UCLA) [39]; rejection sensitivity using the Rejection Sensitivity Questionnaire (RSQ) [40]; and outcome expectation beliefs using the Credibility/Expectancy Questionnaire (CEQ) [41]. We will explore participants' beliefs about their intervention allocation by asking them to guess their assigned treatment condition, rate their confidence in this guess, and provide potential reasons for their guess at weeks 2, 4, and 6 (post-treatment) using a custom-designed questionnaire.
Furthermore, participants will be asked to participate in optional, exploratory MRI assessments prior to treatment (mri_t0; additional MRI sequences will be added to the mandatory safety measurement), within two weeks after the end of treatment (mri_t1), and 3 months after randomization (mri_t2). MRI data will be used to explore potential longitudinal effects of iTBS on brain anatomy and functioning, identify potential moderators of treatment response, and investigate dose–response relationships using electric-field modeling. Imaging sequences will encompass anatomical (such as T1-weighted and T2-weighted structural sequences, diffusion tensor imaging, DTI) and functional scans during resting state (rsfMRI). Participants will receive a reimbursement of 50 € per MRI assessment.
Statistical methods and analysisThis protocol pre-specifies only the primary sequential analyses; others are exploratory. Safety assessments will be descriptive. All analyses will be conducted in R [42]. The analysis scripts are available at https://doi.org/10.17605/OSF.IO/B397U.
Following the methodology of Blackwell et al. [15], we will employ sequential analyses (see Fig. 3) with directional Bayes factors (BFs) to assess the efficacy of active and sham iTBS on the primary outcome. BFs [43] essentially calculate the probability of the observed data under the alternative hypothesis (active iTBS reduces MADRS scores more than sham) divided by the probability of the observed data under the null hypothesis (no superiority of active iTBS). The progression of the trial depends on pre-defined parameters: the minimal sample size per arm, Nmin, at which sequential analyses are initiated; a maximum sample size, Nmax; a BF threshold indicating lack of superiority, BFfail; and a BF threshold indicating superiority, BFsuccess. The specific parameter set was determined by simulation (see below). The trial will halt upon reaching any threshold or Nmax. In the latter case, discussions on extending recruitment may occur with the funder, pending additional ethical approval. Participants ongoing in treatment at trial cessation will complete it, with their data included in final analyses but not in further sequential analyses.
Fig. 3
Sequential Bayesian analysis design
Analyses will follow an intention-to-treat approach, including all participants randomized to a condition, irrespective of intervention completion or outcome data availability. For sensitivity analyses, participants will be classified as per protocol if they complete the 6-week treatment period and receive at least 20 treatment sessions within six weeks. Sequential analyses will commence once each arm meets the predefined minimum sample size Nmin. Analyses will be updated with each participant’s trial completion. Following Blackwell et al. (2022), missing data will be handled using constrained longitudinal data analysis (cLDA) via the R package nlme [44]. Bayes factors will be estimated with the R package BayesFactor [45] using the t-statistic for the Time x Group interaction and directional default Cauchy prior (rscale parameter = √2/2). Effect sizes (adjusted Cohen's d) and estimated means for the primary outcome, along with their 95% confidence intervals (CIs), will be derived from the cLDA model using the R packages effectsize [46] and emmeans [47]. Analyses will be performed by a statistician (SEB) aware of participant allocation (given the directional nature of the analyses), sharing results with the research team only if a BF boundary is met or after Nmax is reached.
Sample size calculationUsing pairwise comparison simulations, we set analysis parameters to achieve a false-positive rate of < 5% and a power of ~ 80% for detecting an effect size of d = 0.6. The choice of effect size was revised after the trial commenced but prior to the start of sequential analysis, based on a recent meta-analysis of iTBS [48]. Initially, we selected an effect-size of 0.8 due to practical considerations, including the absence of specific evidence for iTBS in younger populations and constraints on trial funding. For Nmin = 26; Nmax = 45; BFfail = 1/5; and BFsuccess = 5, simulations with up to 15% missing data suggest that these parameters provide a < 5% false-positive rate and 71.35% power to reach the BFfail threshold before recruitment of Nmax when d = 0; and 81% power to hit BFsuccess before Nmax for an effect size of d = 0.6(see Table 2). For practical purposes, Nmax will be operationalized on the basis of the total sample size, that is, when the total number of participants randomized is 2 × Nmax (i.e., N = 90), regardless of the distribution across arms. Simulation scripts and results of the initial and revised sample size calculations are available at https://doi.org/10.17605/OSF.IO/B397U.
Table 2 Pairwise comparison simulations using cLDASimulations for the following parameters: Nmin = 26, Nmax = 45, BFfail = 1/5, BFsuccess = 5; 15% missing data. Simulations were conducted in RStudio running R version 4.2.2, using 2000 simulations. Estimated effect sizes (d) are a form of Cohen’s d estimated from a t-test on the change scores between groups, with positive d values indicating superiority of the treatment condition to the sham condition. Although d values at intervals of 0.1 were simulated (e.g. 0.2, 0.3, 0.4), the table presents the mean observed effect size across the simulations, which often deviate slightly from these values.
Patient and public involvementA patient and public representative (PB) has reviewed the patient information and consent documents, contributed to this manuscript, and will participate in recruitment, safety monitoring, interpretation of results, dissemination of results, and writing of the final report.
Ethical approval and disseminationThe Ethics Committee of the Medical Faculty of the Ludwig Maximilian University Munich approved the first version of the study protocol (version 1.0) on 09.06.2023 and the latest version (version 1.2) prior to the start of recruitment on 22.03.2024. An amendment of the protocol containing revised sample size calculations is currently prepared for submission to the Ethics Committee. The trial has been registered at the German Clinical Trials Register (https://www.drks.de/drks_web/; DRKS00033313). It will be conducted in accordance with the Declaration of Helsinki and GCP-ICH guidelines. The study's results will be published regardless of the outcome.
Trial statusRecruitment of participants started on 01.04.2024. As of the submission date of this manuscript, one participant has been enrolled in the trial.
留言 (0)