
記住我







We report on a complex clinical and imaging phenotype, including a neuronal migration defect, SBH, in a child harboring a newly discovered compound heterozygous pathogenic variant in the CCDC88A gene. The phenotype includes profound global developmental delay, congenital microcephaly, dysmorphic features, hypotonia, and epileptic encephalopathy.
Biallelic truncating variants in CCDC88A have been described in patients with progressive encephalopathy with edema, hypsarrhythmia, and optic atrophy (PEHO-like syndrome, MIM 617507). This type of EE caused by CCDC88A pathogenic variants is extremely rare. To date, only three reports have been published on this condition: one by Nahorski et al. [18], which included three patients from the same family, one by Abdulkareem et al. (2018) describing two siblings, and, recently, one child described by Issa et al. [10] (Table 1). All the children were born from consanguineous parents. Many clinical features were shared by all the reported patients, such as microcephaly, neonatal hypotonia, seizures, profound developmental delay, face and limb edema, and dysmorphic features, with a similar appearance of eyes, nose, mouth, and fingers (Tables 1 and 2). Regarding visual impairment, all patients exhibited poor or absent visual fixation, and optical atrophy was observed in five of the six patients. Our patient’s phenotype overlaps with most of these clinical traits, except for edema and optic nerve atrophy. EEG studies revealed hypsarrhythmia in the patients reported by Nahorsky et al. (2016) and Issa et al. [10], as well as in our patient.
Table 1 Clinical characteristics and genetic findings of our patient in comparison with previously reported patients with CCDC88A variantsVarious brain malformations have been described in patients with CCDC88A variants. All three cases reported by Nahorski et al. [18] included pachygyria, polymicrogyria prominent in the Sylvian fissures, dilated ventricles, hypoplastic corpus callosum, subependymal cysts, and hypoplastic pons. The patient described by Issa et al. [10] also presented with MRI abnormalities, such as abnormal gyration with minimal augmentation of the cortical thickness, dilated ventricles, hypogenesis of the corpus callosum, demyelination, colpocephaly, and prominent basal ganglia. For the two siblings reported by Abdulkareem et al. (2018), brain atrophy was the only MRI feature described.
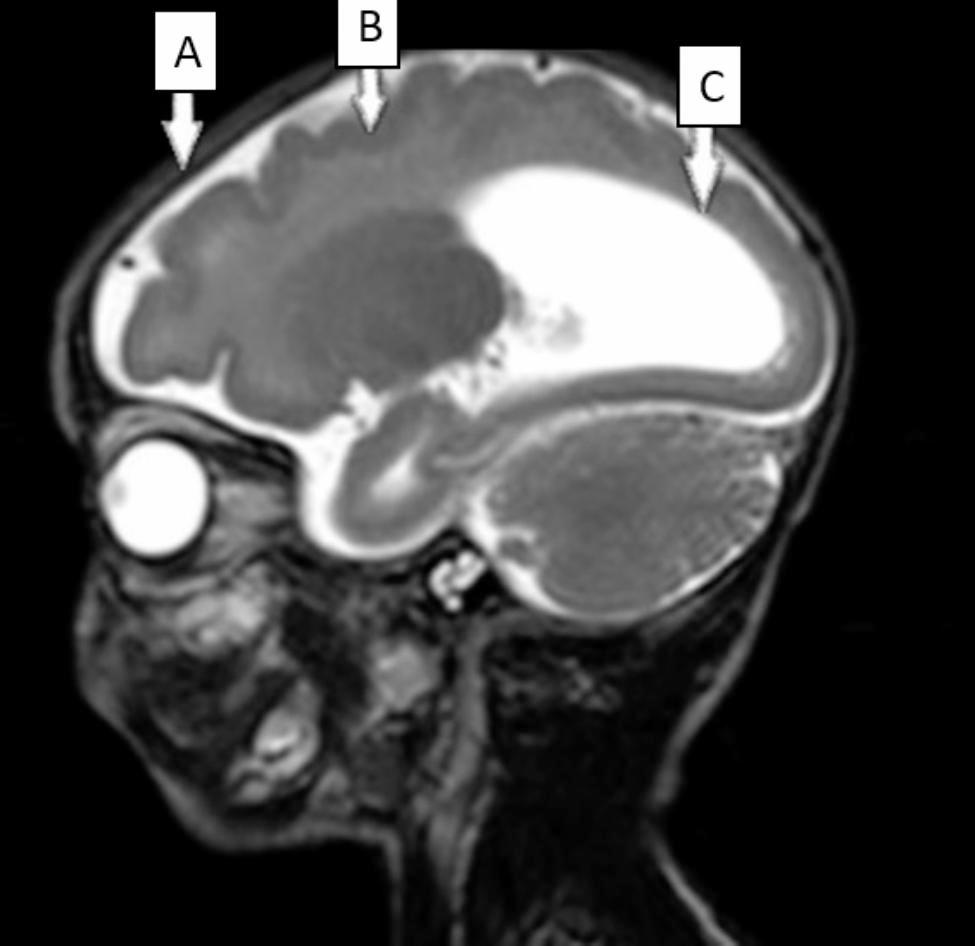
Table 2 Characteristics of the epileptic seizures in our patient and previously reported patients with CCDC88A variantsMRI of our patient showed brain malformations that were consistent in some of those previously reported, with the overlapping features including corpus callosum anomalies, abnormal gyration patterns, and colpocephaly. In addition, our patient presented with subcortical band heterotopia, a severe brain malformation that, until the present time, was not described in patients with PEHO-like syndrome and CCDC88A pathogenic variants.
The differential diagnosis of PEHO-like syndrome should include all types of developmental and epileptic encephalopathies with neonatal onset, such as early-infantile developmental and epileptic encephalopathy and infantile epileptic spasms syndrome. In most of the cases the etiology is genetic, with a wide range of genetic changes reported, from pathogenic sequence variants in genes such as SCN2A, SCN8A, KCNQ2, TSC1, TSC2, ARX, STXBP1, CDKL5 to entire chromosome anomalies (e.g. trisomy 21). However, non-genetic causes should also be excluded, with emphasis on congenital infections (excluded based on specific blood tests for TORCH infections), metabolic conditions (no brain malformations are present on MRI), hypoxic ischemic encephalopathy (excluded based on child history for hypoxia at birth and on specific brain MRI lesions) [29]. The diagrammatic representation of a proposed diagnostic workflow can be found in Fig. 3.
Fig. 3
Diagnostic workflow of developmental and epileptic encephalopathies with early onset
The severity of the clinical findings observed in all the patients indicates the biological significance of CCDC88A, specifically in brain development. The CCDC88A gene encodes a protein, also known as Girdin, which is involved in various biological processes, such as cell, organ, and embryo development [3, 11, 23, 28], neuronal and tumoral cell migration [4, 6, 8, 9, 12, 20, 22, 27], and cancer cells invasion and metastasis [5, 7, 12]. Girdin is expressed across all human tissues, with the highest level being recorded in the brain and testis [4, 17].
The Girdin protein encompasses 1870 aa and has a complex domain architecture that comprises an N-terminal (NT) end represented by a microtubule-binding Hook domain that binds DISC1 (1-196 aa) [24], a coiled-coil domain (196–1304 aa) that plays a role in homodimerization [4, 5], and a terminal (CT) region that includes domains and sites relevant to Girdin’s function. The first of the CT domains, the Gά-binding domain (GBD) (1343–1424 aa), encompasses a PI4P-binding site (1390–1408), which mediates interactions with the plasma membrane and Golgi apparatus, as well as other possible unknown functions. Next to the PI4P-binding site stands the phosphorylation site of AKT serine/threonine kinase 1 (AKT1) (Serine 1417). The terminal part of the CT region contains amino acids 1623–1870 and is responsible for interactions with the actin filaments, AKT1, and epidermal growth factor receptor [4, 8].
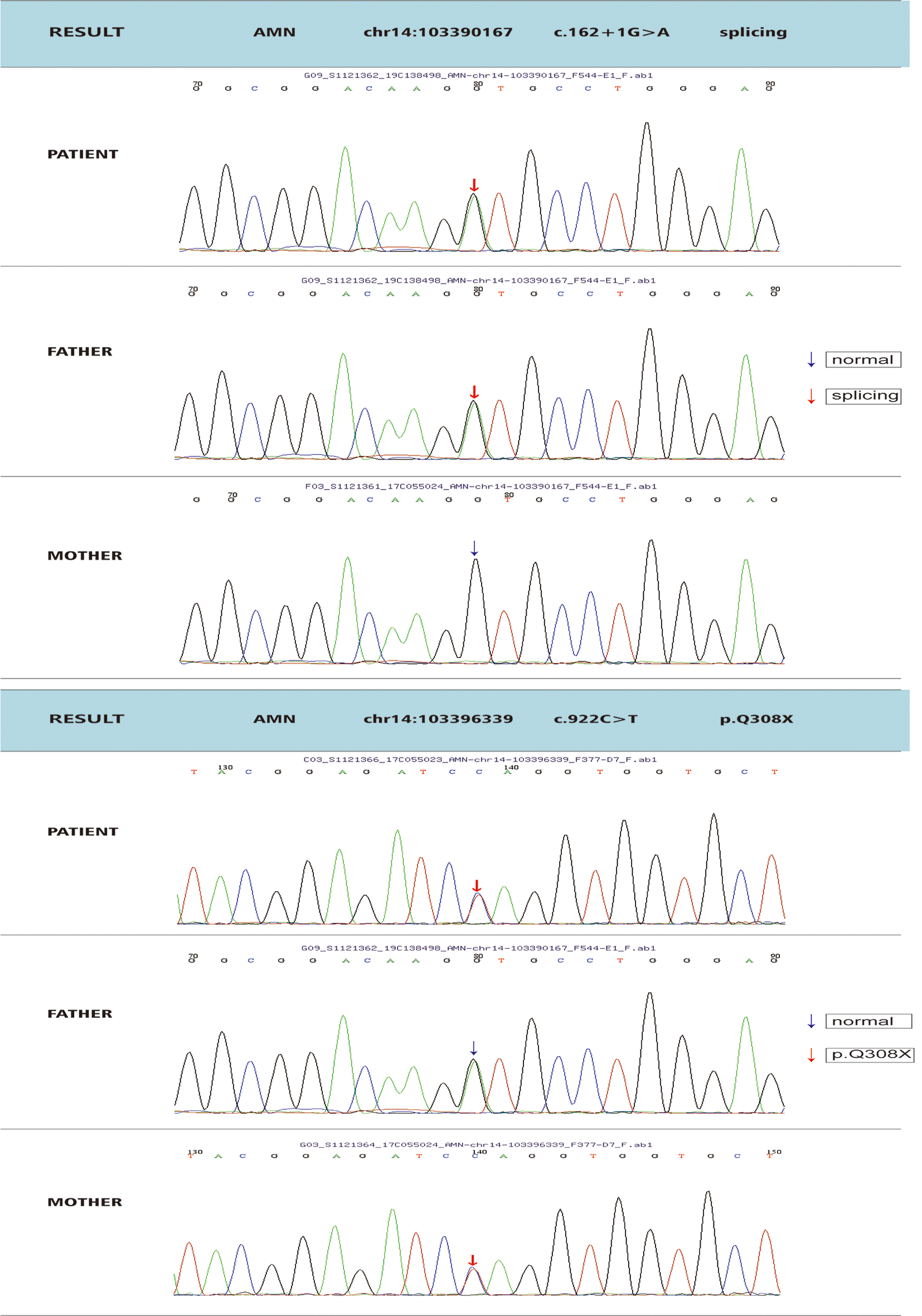
The CCDC88A pathogenic variants described previously [1, 10, 18] are homozygous, truncating variants located in the coiled-coil domain of the protein. Our patient presented with truncating variants in a compound heterozygous pattern, one of which was located in the coiled-coil domain and the other affecting GBD. All of these variants, including the variants in our patient, have been predicted to generate truncated proteins, as they lack the CT domains. Nahorski et al. [18] performed gene expression studies and demonstrated that mRNA containing the variant was present in a proband’s blood sample and was thus not degraded by nonsense-mediated decay. However, the expression level was lower in the patient than in his parents. The production of a truncated protein was found by cloning studies, suggesting that this is the most likely mechanism of the disease [18]. These results further imply that the CT region of CCDC88A is critical for the normal function of Girdin. Mouse studies [2] have shown that Ccdc88a knockout animals exhibit mesiotemporal lobe epilepsy and postnatal growth retardation with early-age lethality. The same results were observed by Nahorski et al. [18], and further analysis of brain anatomy in Ccdc88a knockout mice revealed microcephaly and corpus callosum developmental anomalies, which partially reflected the human brain phenotype.
The above findings indicate that CCDC88A may be a critical gene for normal neurodevelopment that impacts both the function and the anatomical structure of the mammalian brain [16, 18]. Several studies have revealed that Girdin interacts with the Disrupted-In-Schizophrenia 1 protein (DISC1), an important regulator of neuronal migration and differentiation during mammalian brain development from embryonic stages to adulthood [6, 14, 15]. Using curated experimental data from the existing literature and applying a mathematical model (Boolean network), John et al. [13] detailed the DISC1 interactome involved in the regulation of neuronal migration. DISC1 interacts with 18 proteins, which were categorized into eight functional modules. In this system, Girdin, together with AKT1 and actin beta, forms a distinct functional module that mediates the tangential migration of cortical interneurons [13, 25]. It should be noted that in an earlier study, Enomoto et al. [4] proved for the first time that Girdin, an actin-binding protein that is phosphorylated by Akt1, is essential for actin cytoskeleton organization and cell migration. Furthermore, Girdin was found to be involved in the migration of new neurons from the ventricular–subventricular zone of the lateral ventricles to the olfactory bulb, thereby contributing to the development of the postnatal mouse brain [20, 27].
The CCDC88A gene may also play an important role in primary cilia development and function. For instance, Nechipurenko et al. [19] demonstrated that Girdin regulates cilia morphology by positioning the basal body for cilium formation in the sensory neurons of C. elegans and human RPE-1 cells [19]. Future studies investigating the disruption of Girdin might provide new data on its involvement in central nervous system development, thus helping to explain human structural brain anomalies in patients with truncating variants of CCDC88A.
In conclusion, we present a patient with a PEHO-like clinical picture harboring a novel sequence variant of the CCDC88A gene, thus contributing to the phenotypic and genotypic delineation of CCDC88A-related EEs. In addition, our patient presented with a complex brain malformation, including subcortical band heterotopia, which was not reported in previous cases.
留言 (0)