
記住我








Patients in the control group will take an oral placebo that visually and physically matches the investigational product. All study participants will receive standard care (e.g. vaginal progesterone, serial cervical length measurements, etc.).
Intervention descriptionPregnant women at risk for sPTB who are eligible for inclusion will be included and randomised into the intervention or control group at 80/7–106/7 weeks of gestation. All study participants will start taking synbiotics or placebo, respectively, immediately after treatment allocation and continue until delivery. The daily dose is two capsules, one in the morning and one in the evening. Patients will be instructed about normal personal intimate hygiene and advised to avoid excessive genital cleaning or vaginal washings.
The investigational product (IP) is an oral synbiotic containing eight probiotic Lactobacillus strains, in total 2 × 1010 colony-forming units (CFU) per daily dose of two capsules, furthermore the prebiotics inulin, fructooligosaccharids (FOS), and D-mannose; see Table 2. The excipients are magnesium bisglycinate, magnesium stearate, and silicon dioxide. Capsules with enteric coating will be used to ensure delayed release for both the IP and placebo. The placebo capsules will only contain the excipients mentioned above. IP and placebo will be stored below 25°C.
Table 2 Composition of the investigational productCriteria for discontinuing or modifying allocated interventionsPrevious studies demonstrated the safety of pro- and synbiotics in pregnancy [41, 42]. Therefore, we do not anticipate serious adverse events or complications secondary to taking either the IP or the placebo that could necessitate treatment discontinuation or modification.
Strategies to improve adherence to interventionsPatients will be given a diary that includes reporting the intake of the IP or placebo, a visit schedule, and contact information. The diary will be reviewed by a delegated team member and discussed with the patient on every study visit. Adherence to the intervention will be checked based on the patient’s diary entries and the number of returned, unused capsules of IP or placebo.
Relevant concomitant care permitted or prohibited during the trialAt each study visit (including unscheduled visits such as hospital admissions), concomitant medication is checked and documented by the principal investigator (PI) or delegated team member. Because certain interventions may influence the study outcomes, standardised treatment protocols for indications that are relevant for the research question were made in collaboration with all participating centres. Recommendations were made for the use of vaginal progesterone (200 mg once daily for the indication of sPTB prevention), tocolysis, corticosteroids for foetal lung maturation, magnesium sulphate for neuroprotection, cervical cerclage or pessary placement, antifungal medication for symptomatic Candidiasis, and antibiotics for PPROM, Group B Streptococci (GBS) prophylaxis, symptomatic BV, and common sexually transmitted infections. The use of any pre-, pro-, or synbiotic other than the IP is not allowed during the study.
Provisions for post-trial careN/a. Post-trial care includes the routine postpartum follow-up and this is not influenced by trial participation. We do not anticipate any harm from trial participation.
OutcomesThe primary outcome is the gestational age at delivery in weeks plus days, expressed as mean and standard deviation (SD) for both groups. Secondary outcomes include PTB rates in subcategories based on the GA at delivery (extreme PTB from 240/7 until 276/7 weeks, severe PTB from 280/7 until 316/7 weeks, and moderate to late PTB from 320/7 until 366/7 weeks of gestation) expressed as number (n) and proportion (%), PPROM rates (n, %) and GA at PPROM (weeks + days), vaginal microbiome analysis (see further), midstream urine culture at 16 weeks of gestation, GBS screening at 35 to 37 weeks of gestation, placental pathology, and neonatal outcomes (see further).
The vaginal microbiome will be examined once per trimester in order to correlate the effect of the oral synbiotic on the microbiome with pregnancy duration and duration of intake. Vaginal swabs will be taken at inclusion (80/7 to 106/7 weeks), at 190/7 to 210/7 weeks, at 290/7 to 310/7 weeks, at delivery, and upon admission at the high-risk antenatal ward for threatened preterm birth (PPROM, preterm labour or short cervix). The vaginal microbiome will be analysed by bacterial culture and metataxonomic 16S rRNA gene sequencing. Because of potential interference with NGS analysis, patients on vaginal progesterone for PTB prevention are instructed to hold the dose of progesterone the evening before that study visit and to resume immediately after the study visit. Furthermore, the following swabs will be sampled during the PRIORI trial, frozen, and stored for metataxonomic sequencing with alternative funding: one vaginal swab for NGS at day 0, 3, 6, 9, 14, and 28 as long as the patient is admitted after PPROM, placental swabs in sPTB cases, and neonatal meconium swabs in PPROM cases. The results of the swabs mentioned above, sampled in the context of this study, will stay blind until the end of the trial and will not influence the patient’s care.
During the internal pilot phase of the RCT, additional vaginal swabs will be taken during one extra study visit 4 weeks after the start of treatment. The primary outcomes of the internal pilot study are (1) the difference in total Lactobacillus abundance after 4 weeks of treatment compared to baseline by metataxonomic sequencing and (2) the vaginal detection of the Lactobacillus gasseri strain of the IP after 4 weeks of treatment by quantitative PCR (qPCR). The choice for L. gasseri as qPCR target is based upon the relatively low prevalence of natural L. gasseri dominance (< 10%), as compared to L. crispatus (around 40%) [43], in our European patient population (recently confirmed in the large-scale Belgian Isala project, Antwerp University), and the technical limitations in strain-specificity of qPCR.
Significant differences in pregnancy duration may be reflected in improved neonatal outcomes. Based on the Delphi consensus and in line with the Core Outcome Measures in Effectiveness Trials (COMET) initiative [44], we selected the following neonatal outcomes: neonatal mortality, birth weight, necrotising enterocolitis, bronchopulmonary dysplasia, intraventricular haemorrhage, encephalopathy of prematurity, infectious parameters (duration and number of antibiotic courses, early and late-onset sepsis), duration and type of respiratory support, surfactant administration, retinopathy, other neonatal morbidity, and duration of neonatal admission.
The economic impact and quality of life (QoL) will be assessed using the Work Productivity and Activity Impairment (WPAI) and EQ-5D questionnaire, respectively, during visit 2, 3, 4, 5, unscheduled visits (at admission and after 1 week), at delivery and during the neonatal follow-up period.
For continuous variables, mean and SD will be presented by study group and the difference (treatment effect) will also be presented with a 95% confidence interval. For binary variables, counts and percentages will be presented by the study group. The odds ratio, comparing the intervention group with the control group, and 95% confidence interval will also be presented.
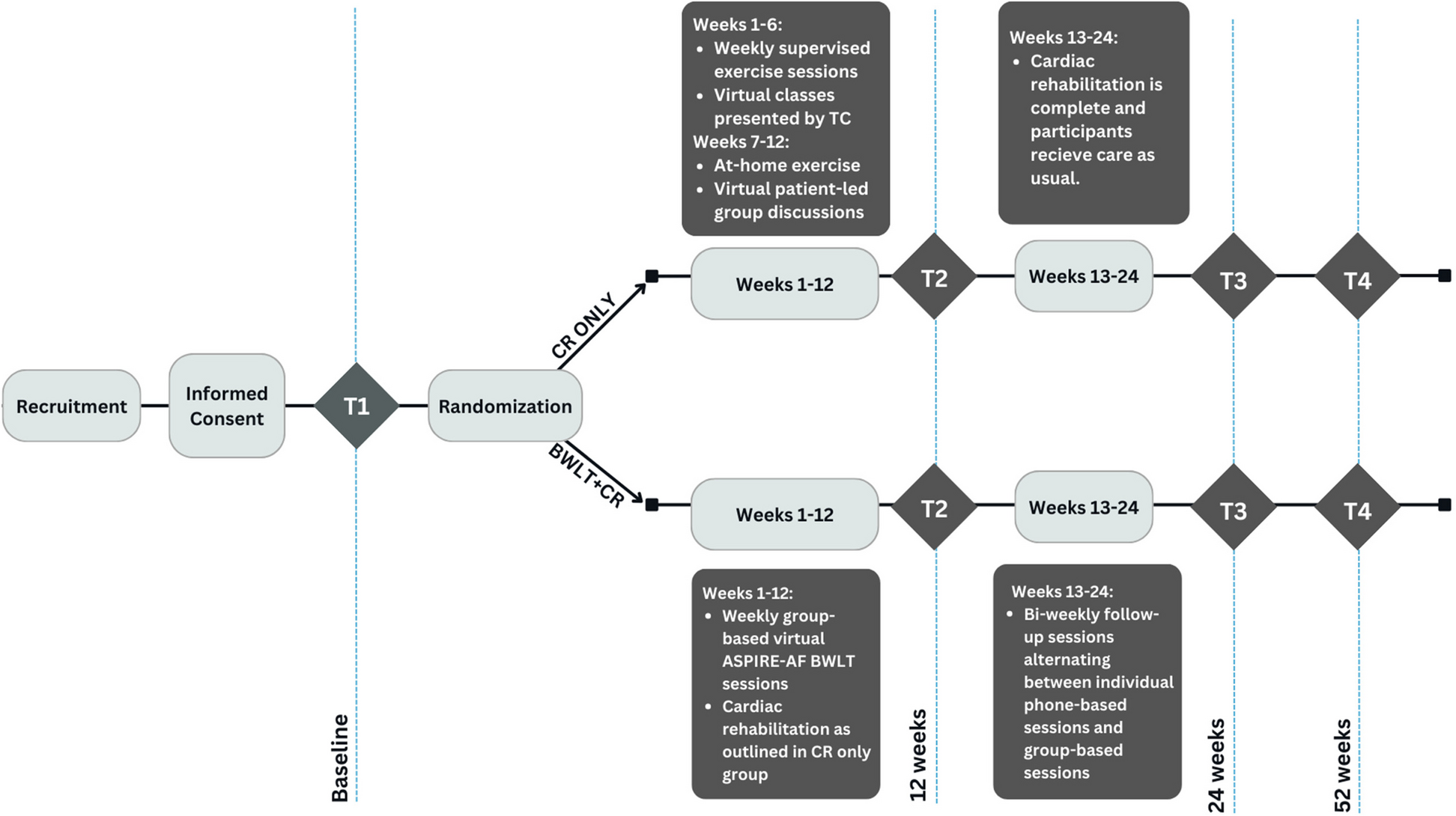
Participant timelineThe schedule of enrolment, interventions, and assessments can be found in the schematic diagram in Fig. 1.
Fig. 1
Schedule of enrolment, interventions, and assessments. 1 Visit number and gestational age in weeks (w). 2 Admission on the high-risk antenatal ward for preterm labour, PPROM or short cervix. 3 New informed consent forms will be signed by the mother for the collection of neonatal data. 4 Height is only measured on visit 1 to calculate start Body Mass Index. 5 Microbial culture and metataxonomic sequencing. 6 Transvaginal ultrasound (TVUS). 7 Group B Streptococci (GBS) rectovaginal swab. 8 Quality of life questionnaire. 9 Work Productivity and Activity Impairment questionnaire
Sample sizeThe sample size calculation is derived from the primary outcome: gestational age at delivery (continuous variable). To detect a clinically relevant difference in pregnancy duration of 1 week between the intervention and control group with sufficient statistical power (i.e. 90%), assuming a SD of 3 weeks [27, 28], and with an alpha of 0.05, 382 patients are required in a 1:1 randomisation. These power calculations were based on a two-sided two-sample t-test. Information regarding the correlation between patients from the same centre, quantified by the intraclass correlation coefficient, is currently unavailable and thus could not be factored into the sample size calculation. However, it is anticipated that the intraclass correlation coefficient for gestational age at delivery is small, and therefore, the conducted power calculations are deemed applicable.
Anticipating that the primary outcome may not be available for a small proportion of the randomised patients due to reasons such as lost to follow-up or withdrawal of consent, a dropout rate of 5% is accounted for to maintain a power of 90%. Consequently, the total number of patients to be recruited for the trial is calculated as 402.
The sample size calculation was conducted using SAS for Windows, version 9.4.
RecruitmentPatients will be recruited in seven Belgian teaching hospitals within a period of approximately 36 months. Depending on the recruitment speed, more sites will be activated to enrol 402 participants. The initial approach for pre-screening potential patients will be done by a member of the patient’s existing clinical care team. Only physicians who are members of the PRIORI study team will inform the patient. If the treating physician is not a member of the PRIORI study team, he or she could refer the patient to a PRIORI investigator. The PRIORI investigator will confirm the eligibility of the patient and then the screening process can be started after a written informed consent is obtained.
留言 (0)