
記住我


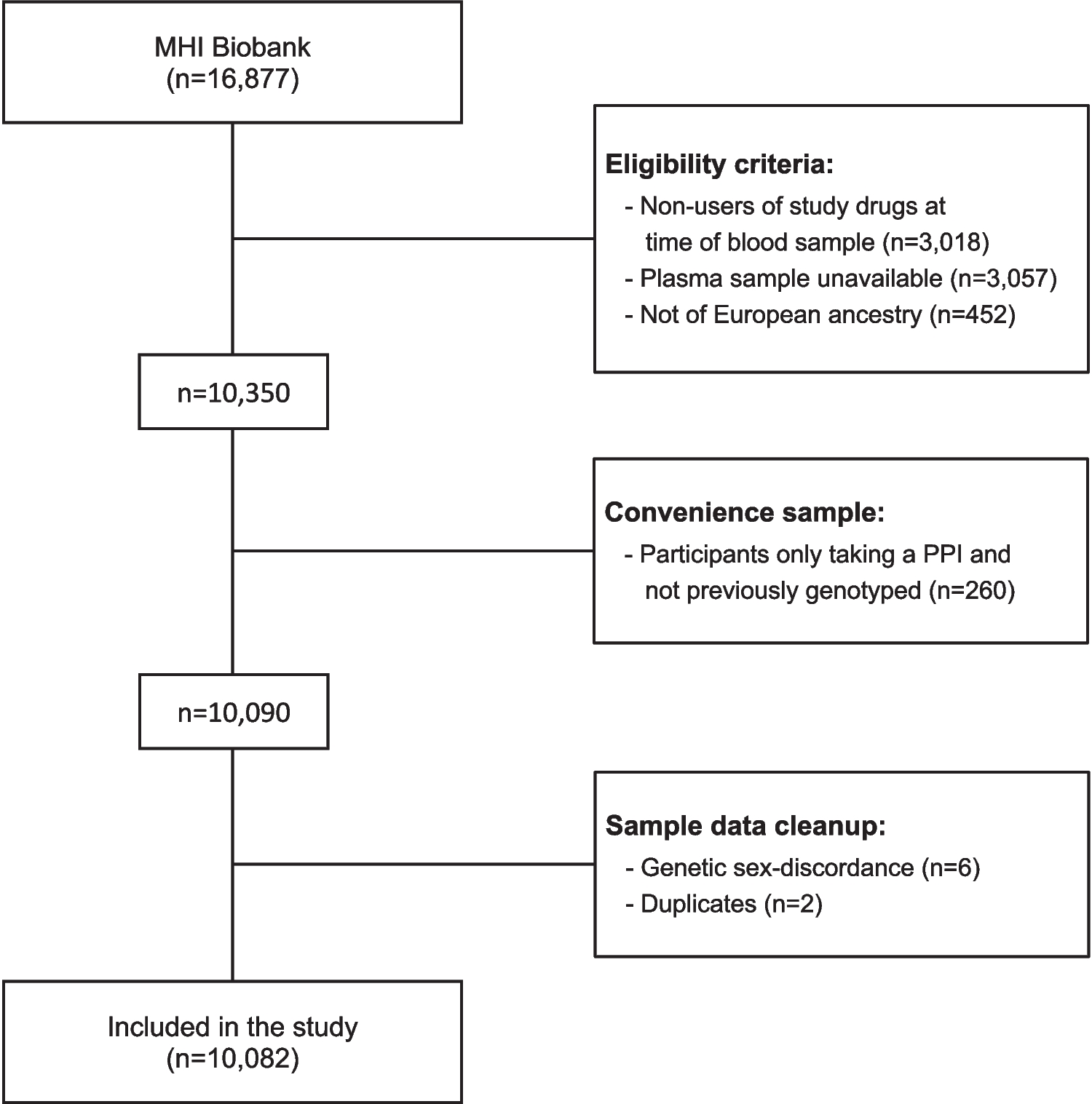




A total of 211 patients aged between 0.1 and 63 years were recruited from two sites in New South Wales, Australia. The study was a prospective investigation on the pharmacokinetics of F-Ara-A in patients ranging from neonates to adults who were administered fludarabine as part of HSCT conditioning. Participants were recruited either to a multi-centre clinical trial entitled “Pharmacokinetics, pharmacodynamics and pharmacogenomics of busulfan and other agents used in blood or marrow transplantation”, registered with the Australian Clinical Trials Registry (Registration number: ACTRN12612000544875, registration date: 22/05/2012) or an earlier single-centre study “Pharmacokinetics of fludarabine des phosphate in children receiving fludarabine”. The study protocols were approved by the Sydney’s Children’s Hospital Network Human Research Ethics Committee, and written consent was obtained from parents/guardians of children and/or participants.
Fludarabine dose administrationThe patients in this study were treated according to specific BMT conditioning protocols which varied with respect to the total administered fludarabine dose and the number of days over which it was administered (range, 3 to 6 days). Fludarabine was infused over 0.42 to 1.5 h (median 0.5 h). The BMT conditioning protocols used combinations that contained busulfan in 155 transplants (73%) and treosulfan in 35 transplants (17%) (Table 1); in these protocols, fludarabine was administered immediately prior to the busulfan or treosulfan infusions. A surface area-based fludarabine dose (range, 10 to 50 mg/m2) was administered in 194 transplants, whilst a weight-based dose of 1 to 2 mg/kg was used in 18 transplants where the patients weighed < 11 kg. In two patients with GFR values of 41 and 30 mL/min/1.73 m2, respectively, the initial fludarabine dose was reduced by 50%. Real-time F-Ara-A pharmacokinetic assessment based on total F-Ara-A concentrations was conducted in two patients, a child with a GFR of 30 mL/min/1.73 m2 and a pre-term baby transplanted at term, and this led to further fludarabine dose reductions following a reduced initial dose.
Table 1 Clinical data of 211 patients who received fludarabine as part of allogeneic HSCT conditioningBlood sampling for F-Ara-A pharmacokinetic analysisA series of five to eight blood samples were collected at timed intervals after the first, second, third, fourth and fifth fludarabine doses. In 155 patients who received busulfan, fludarabine was infused immediately prior to the busulfan infusion, with blood for fludarabine concentration assessment collected at the end of the fludarabine infusion then at 0 h, 1 h, 2 h, 4 h and 8 h after the busulfan infusion (1.5- to 3-h duration). Similarly, in 35 patients who received treosulfan, fludarabine was infused immediately prior to the treosulfan infusion (2-h duration), with blood collection at the end of the fludarabine infusion (then at 0 h, 0.5 h, 1 h, 3 h, 5 h, 7 h and 12 h after the treosulfan infusion). In those children who did not receive busulfan or treosulfan, blood sampling was at 0 h, 0.25 h, 0.5 h, 1 h, 3 h, 6 h and 12 h after the end of the fludarabine infusion. A total of 74 patients also had samples collected from between 12 and 24 h after the dose. Bloods were centrifuged for 10 min at room temperature at 3400 rpm within 60 min of collection and the plasma was stored at − 40 °C until analysis.
F-Ara-A concentration measurementThe clinical pharmacokinetics of fludarabine was evaluated by studying the total and unbound concentrations of the primary metabolite, F-Ara-A. Detection and quantification of total and unbound F-Ara-A were carried out using a Shimadzu Nexera ultra-high-performance liquid chromatography (UHPLC) system fitted with two LC-30AD pumps, a SIL-30AC autosampler and a SPD-M30A photodiode array detector set at 262 nm. For determination of total plasma concentration of F-Ara-A, 100-µL plasma aliquots from patient samples and spiked standards (0.1, 0.2, 0.5, 0.75, 1, 2, 5 µg/mL) were deproteinized by addition of 25% trichloroacetic acid (50 µL) and water (150 µL). After vortex mixing and centrifuging at 13,000 rpm, a 20-µL aliquot of the supernatant was injected into the UHPLC system. For determination of unbound F-Ara-A concentration, 200-µL aliquots of patient plasma samples were deproteinized by rapid ultrafiltration with Amicon 0.5-mL ultracentrifugal filters (PN: UFC501096) that utilized regenerated cellulose with a molecular weight cut-off of 10,000 units. Calibration was against spiked standards (0.1–5 µg/mL) prepared in water, a similar aqueous matrix as plasma ultrafiltrate. HPLC separation was achieved using a Kinetex XB C18 2.9 µm 100 × 4.6 mm column (Phenomenex 00D-4496-E0) fitted with a C18 4.6 mm ultra security guard (Phenomenex: AJO-8768) using an isocratic mobile phase that consisted of 50 mM sodium phosphate buffer, pH 4, with 9% acetonitrile and 0.1% sodium octyl sulphonate (10 g/100 mL). Using a flow rate of 1 mL/min, the retention time of F-Ara-A was 4.3 min. The total F-Ara-A assay was linear from 0.1 to 5 µg/mL with acceptable precision (< 13% coefficient of variation, CV) and accuracy (bias < 6% deviation from the nominal concentration) for spiked plasma controls, with F-Ara-A concentrations of 0.2, 0.75, 1.5 and 5 µg/mL (n = 66). The limit of quantification (LOQ) for the total F-Ara-A assay was 0.1 µg/mL (CV = 20%, bias = 1%, n = 66). The unbound F-Ara-A assay was linear from 0.1 to 5 µg/mL with acceptable precision (CV < 15%) for the same spiked plasma controls (n = 55). A negative bias was observed, with estimates of 26%, 21%, 16% and 16% recorded for the 0.2, 0.75, 1.5 and 5 µg/mL concentrations, respectively. This may not reflect the true accuracy of the unbound F-Ara-A assay as protein binding may have occurred during the spiking process. The LOQ for the unbound F-Ara-A assay was also 0.1 µg/mL (CV = 13%, bias = − 23%, n = 6).
Pharmacokinetic dataThe pharmacokinetic data that was collected included the date and time of fludarabine dose and the date and time of blood collections for F-Ara-A concentration determination. Since the measured F-Ara-A is different from the administered dose (fludarabine phosphate), an equivalent dose of F-Ara-A was used for modelling. This was calculated as the fludarabine dose multiplied by 0.78, which is equivalent to the ratio of F-Ara-A molecular weight (285 g/mol) to fludarabine phosphate molecular weight (365.2 g/mol).
Other data that was collected included patient or treatment-related factors that had the potential to impact the F-Ara-A pharmacokinetic parameters. Data on patient age, sex, weight and height was collected and allowed calculation of other parameters related to body size including body surface area (BSA), body mass index (BMI) and fat-free mass (FFM). BSA was calculated using the equation of Mosteller et al. [20]. FFM was calculated using published formulas for adults [21] and children [22]. Post-menstrual age (PMA, weeks) was calculated as postnatal age + gestational age (40 weeks in all patients except one pre-term infant for whom gestational age was 34.3 weeks). GFR was determined by measuring the plasma clearance of 43Tc99–diethylenetriaminepentaacetic acid for 181 children and the Schwartz-Lyon equation [23] for two children. In adult patients, GFR was estimated using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation [24]. Other information that was collected included whether or not there was pre-existing liver impairment, HSCT conditioning (busulfan-based, treosulfan-based or other), diagnosis group (malignant, immune deficiency or genetic disease), pre HSCT albumin and total protein concentrations and concomitant medications during the period of fludarabine administration, including ganciclovir, allopurinol, clobazam, clonazepam, amphotericin B, fluconazole and serotherapy.
Population pharmacokinetic modelling of total and unbound F-Ara-A concentrationsPopulation PK modelling was performed using nonlinear mixed effects modelling software (NONMEM, version 7.40) with the Perl-speaks-NONMEM library (version 4.9.0) [25] and Pirana (version 2.9.9) as the graphical user interface [26]. A first-order conditional estimation with interaction (FOCE-I) method was used for approximation. Graphical output and statistical analysis were performed using the ggplots2 [27] and ggstatsplot [28] packages implemented in R (version 4.2.3) and RStudio (version 2023.03.0 + 386) [29].
Since a plot of unbound versus total F-Ara-A concentrations (Cunbound, Ctotal, respectively) demonstrated a linear association (Fig. 1), a linear protein binding model was implemented for F-Ara-A. Cunbound and Ctotal were modelled simultaneously with Cunbound as the central compartment. Ctotal was predicted from Cunbound and fraction unbound (fu) (Eq. 1):
Fig. 1
Scatterplot of unbound versus total F-Ara-A concentrations from 22 patients
The basic structural model was selected by comparing the performance of the two- and three-compartment pharmacokinetic models, assuming first-order kinetics. A one-compartment model was not tested since visual inspection of the concentration versus time plots indicated multi-phase elimination profiles. Inter-individual variability (IIV) was assumed to follow a log-normal distribution. Inter-occasion variability (IOV) was implemented similarly, with each dose defined as a separate occasion. Residual error was evaluated as either proportional, additive or a combination of the proportional and additive error models. Potential predictors of variability in the pharmacokinetic parameters were screened as covariates for inclusion in the model. Continuous covariates were evaluated using a linear or power function described in Eq. 2:
$$_}=_} }_}/}_}\right)}^},$$
(2)
where Covi is the covariate value for the ith individual, Covtypical is the typical or median value in the population and k is the exponent which was fixed at 1 for a linear function or estimated for a power function. Binary categorical covariates were evaluated using Eq. 3:
$$_}=_}\left(1+_}\right),$$
(3)
where Pcov is the estimated proportional factor with which the parameter changes for a specific covariate.
Renal function was incorporated into the model by differentiating total unbound clearance (CLu) into non-renal and renal components as previously described [12]. Body weight was applied to total CLu as previously described [12]. Alternative descriptors of body size (FFM, BSA, BMI, height) were evaluated as a replacement for weight. Height, BSA and BMI were scaled linearly. Allometric scaling factors of 0.75 for unbound CLu parameters and 1.0 for the volume of distribution (Vu) parameters were applied to the weight and FFM covariates, with the weight covariate centred using a reference body weight of 70 kg. The impact of patient maturation (FMAT) was separately evaluated on renal and non-renal clearance using a standard maturational model [30] that incorporated PMA, a sigmoid function (HILL) and the PMA representing 50% renal maturation (PMA50):
$$FMAT=\frac^}}}^}+^}}$$
(4)
The Hill exponent was restricted to be less than 5 in order to obtain a plausible maturation curve.
Stepwise forward selection and backward elimination methods using the likelihood-ratio test were employed to determine covariate significance, with covariates selected for inclusion if they decreased the model objective function value (OBV) by more than 3.84 units (p < 0.05) during forward selection and 6.63 units (p < 0.01) during backward elimination. The most appropriate covariate model was selected based on (1) a low value for the objective function (OBV), (2) low estimates for sigma, (3) low relative standard error estimates for all pharmacokinetic parameters, (4) low estimates of IIV and IOV, (5) low shrinkage of below 30% [31] for the random and residual error components and (6) good model performance as assessed using goodness-of-fit plots, a visual predictive check (n = 1000) and non-parametric bootstrapping (n = 1000).
Derived pharmacokinetic parametersA number of pharmacokinetic parameters for F-Ara-A were derived from the post hoc Bayesian estimates of the primary pharmacokinetic parameters to allow comparison across age groups and GFR categories. CLu and V1u were normalised to WT (70 kg) by dividing by WT and multiplying by 70. Unbound AUC0-∞ (AUCu0-∞) was determined by dividing the dose (mg) by the individual posterior Bayesian estimates of F-Ara-A CLu. Total AUC0-∞ (AUCt0-∞) was calculated as AUCu0-∞ divided by fu. Median and interquartile range for AUCu0-∞ and AUCt0-∞ were then calculated for mg/m2 doses ranging from 10 to 50 mg/m2 to provide an indication of the expected range of exposures using current oncology protocols. Percent unbound F-Ara-A was calculated as fu multiplied by 100, and percent protein bound F-Ara-A was calculated as (1-fu) multiplied by 100.
In order to compare pharmacokinetic results from previously published studies, clearance and volume of distribution parameters were converted to weight- normalised estimates (70 kg) to allow comparison using common units. In addition, total and unbound AUC0-∞ were normalised to mg fludarabine dose and allometric size using Eq. 5 and data on mg/m2 fludarabine dose and the median values of BSA and WT in the populations studied. For those studies in which median BSA and/or WT were not reported, values of 1.73m2 and 70 kg, respectively, were used in the calculations. Linear pharmacokinetics for fludarabine was demonstrated in early studies [32, 33].
$$}_=\frac}_}\;\left(\text\right)}\times \left(\mathrm\right)}\right)}^.$$
(5)
Impact of age on derived pharmacokinetic parameters for total and unbound F-Ara-APatients were sub-divided into groups according to age (< 1 year (n = 29), 1–7 years (n = 69), 7–12 years (n = 38), 12–20 years (n = 48), > 20 years (n = 27)). GFR (mL/min/1.73 m2), F-Ara-A fu, CL(L/h/70 kg), V1 (L/70 kg) and normalised AUCu0-∞ were then compared using the Kruskal–Wallis H test with post hoc pairwise comparison using the Holm-adjusted Dunns test. Violin plots with super-imposed boxplots with statistical comparisons were implemented in RStudio using the ggstatsplot package [28].
Evaluation of F-Ara-A exposure and dosing in renal impairmentThe Kidney Disease Improving Global Outcomes (KDIGO) 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease (CKD) [34] categories were applied to the F-Ara-A AUCu0-∞ and CLu values, as recommended in the International Consensus Guidelines on Anticancer Drug Dosing in Kidney Dysfunction (ADDIKD) [18]. These categories were GFR ≥ 90 mL/min/1.73 m2 (G1, normal or high GFR), GFR 60–89 mL/min/1.73 m2 (G2, mildly decreased GFR), GFR 45–59 mL/min/1.73 m2 (G3A, mildly-moderately decreased GFR) and GFR 30–44 mL/min/1.73 m2 (G3B, moderately-severely decreased GFR). Percent difference of median CLu (L/h/70 kg) for the G2, G3A and G3B categories when compared with the G1 category was then calculated using Eq. 6:
$$\mathrm=\frac\left(\mathrm\right)-\mathrm(\mathrm)}\left(\mathrm\right)}\times 100$$
(6)
In nine individuals with GFR < 60 mL/min/1.73 m2, the mg fludarabine dose required to target the median AUCu0-∞ observed for the relevant mg/m2 dose in the treatment protocol was calculated using Eq. 7 and compared to the mg fludarabine dose calculated using BSA-based dosing. A percent difference in dose within 20–25% reflects the attainment of the target AUCu0-∞ within acceptable limits, the 80–125% interval [35, 36].
$$\mathrm\left(\mathrm\right)\mathrm_-\infty }=\mathrm\left(\mathrm\right)\mathrm\times \frac_-\infty } }_-\infty }}$$
(7)
留言 (0)