
記住我

A previously healthy 14-month-old Hispanic female presented with eight months of worsening rash, weakness, periorbital edema, intermittent fevers, and weight loss. Symptoms started with cough, congestion, and fever at approximately six months of age. She was treated by her primary care provider (PCP) with a five-day course of prednisolone. After completing the course of steroids, she developed a rash on her left arm. The rash persisted despite treatment with moisturizers. The patient then received her one-year-old vaccines (six months following her initial presentation) with subsequent spreading of her rash. Her rash was described as “bruise-like” and spread to include all four extremities along with truncal involvement. Her activity level declined, and she appeared weaker to family members. Six weeks after vaccines (and two weeks prior to presentation for admission to hospital) she began to have daily fevers. She was evaluated again by her PCP four days prior to hospital presentation with lab work obtained showing elevated transaminases, creatine kinase (CK), alkaline phosphatase, gamma-glutamyl transpeptidase (GGT), lactate dehydrogenase (LDH) and a positive antinuclear antibody and ribonucleoprotein antibody. She was subsequently referred to pediatric rheumatology for urgent evaluation.
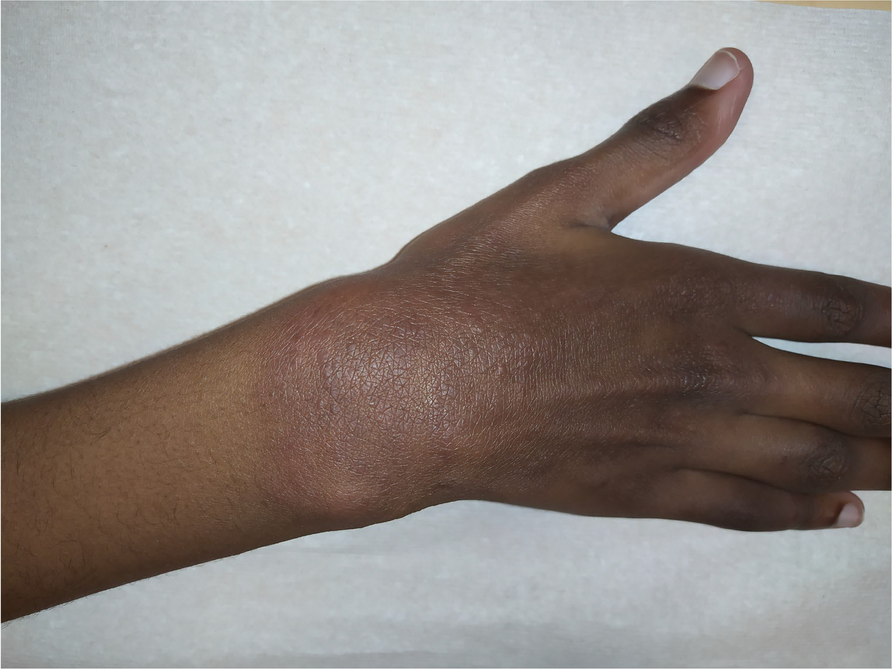
On the day of presentation to the rheumatology clinic, the patient was noted to be thin with her weight in the fifth percentile at 7.9 kg. Her exam was notable for thinning of hair, heliotrope rash, periorbital edema, and violaceous macules on her bilateral elbows, forearms, arms, and knees (Fig. 1). Many of the violaceous macules were also ulcerative, progressing throughout admission. The upper extremity exam was notable for bilateral and symmetric Gottron’s sign and nail fold erythema (Fig. 2). She also had arthritis along with an ulcerative lesion on her left ankle (Fig. 3). The abdominal exam was notable for hepatosplenomegaly. Nail fold capillaroscopy was moderately abnormal. The patient was admitted to expedite her work up with a broad differential including juvenile dermatomyositis, an overlap syndrome, and autoinflammatory syndromes.
Fig. 1
Ulcerative violaceous macules of R elbow, L arm
Fig. 2
Symmetric ulcerative Gottron’s sign and nail fold erythema
Fig. 3
Ulcerative lesion of L ankle
Pertinent positive initial lab work included a respiratory viral panel positive for parainfluenza, elevated transaminases (with normal INR) CK, LDH, aldolase, and bilirubin. A myositis antibody panel including anti-MDA5 was drawn on admission as well. Genetics was consulted and targeted exome sequencing was ordered due to suspicion for syndromic disease based on a broad forehead, triangular facies, protruding ears, smooth philtrum, and thin upper lip.
Initial imaging was notable for mild right ventricular hypertrophy on echocardiogram with previously undiagnosed atrial septal defect, gallbladder sludge, and liver span of 10 cm (upper limit of normal) on abdominal ultrasound, bilateral hip, and thigh myositis with inguinal lymphadenopathy on MRI pelvis, and patchy areas of consolidation and ground glass opacities of bilateral lower lobes on CT chest.
Following the initial diagnostic workup, the patient continued to fever daily for the first ten days of admission. Her rash evolved to include ulceration of bilateral knees, elbows, and metacarpals. While her respiratory status was initially stable, supplemental oxygen was needed due to hypoxia on hospital day three and she continued to have an oxygen requirement throughout the hospital course.
Though findings of the MRI pelvis and CT chest were suggestive of dermatomyositis, immunosuppressive treatment was not immediately initiated due to the need to rule out infectious causes in the setting of two weeks of fever. Further infectious workup was negative over the coming days including testing for Histoplasma, HIV, EBV, viral hepatitis, and tuberculosis.
Additionally, muscle and liver biopsies were obtained on hospital day five. On hospital day eight, a liver biopsy resulted positive for nonspecific mixed inflammatory infiltrate (Fig. 4a-c) and areas of ductopenia and ductular proliferation (Fig. 4d). The patient was also treated with a five-day course of vancomycin and cefepime for hospital acquired pneumonia starting on hospital day eight, but fever curve and oxygen requirement were unchanged over the following three days.
Fig. 4
a-d. Liver histopathology, a Liver core, (H&E 100X). The liver core demonstrates hepatocellular damage with feathering and ballooning of hepatocytes in the lobules. b Liver core, (H&E 200X). The portal area demonstrates mild portal inflammation including eosinophils and rare neutrophils, ductular proliferation, and ductopenia. c Liver core, (PAS 200X). Significant glycogen depletion in the hepatocytes. d Liver core, (CK-7 200X). Cytokeratin-7 immunohistochemical stain showing prominent ductular proliferation at the periphery of the portal area, and absence of the central duct (ductopenia)
On hospital day 11, preliminary results of the muscle biopsy were positive for dermatomyositis with MHC staining pending. Methylprednisolone was initiated at 30 mg/kg daily for three days. The patient responded well, particularly her cutaneous features, strength, and overall energy, though oxygen requirement persisted. IVIG 2 g/kg was given on hospital day 14 and steroid dosing was switched to 2 mg/kg/day. On hospital day 16, anti-MDA5 antibody results returned positive. After IVIG treatment, cyclophosphamide was initiated at 500 mg/m2 on hospital day 19. The patient’s clinical status gradually improved over the next two weeks, and she was discharged on room air though she did require honey thickened formula feeds at home due to video fluoroscopic swallow study showing signs of aspiration near time of discharge while transitioning from nasogastric to oral feeding. Liver function tests gradually returned to normal over time without evidence of chronic liver disease (Table 1).
Table 1 Liver function tests throughout hospitalization (normal range in parentheses)Of note, her genetic evaluation did not reveal any syndromic pathology specific to the patient’s presentation but did return positive for three variants of unknown significance (ERBIN, RFT1, RRM2B) along with known pathogenic variant TMEM67 – causative for ciliopathies but thought to be non-contributory to patient’s initial presentation. Final muscle biopsy results were consistent with dermatomyositis as well as features of congenital LMNA myositis seen in various congenital muscular dystrophies though genetic analysis in our patient did not show any LMNA mutations.
After induction with six months of cyclophosphamide, IVIG infusions, and decreasing frequency of methylprednisolone pulses, our patient was started on maintenance tofacitinib in the outpatient setting. JAK inhibitor therapy with tofacitinib was selected based on the patient's significant ulcerative skin disease, the presence of anti-MDA5, and recent reports of patients being recalcitrant to standard therapy [11]. Her initial dose started at 3.2 mg twice daily but one month into treatment after facial rash was observed and tofacitinib dose was increased to 5 mg twice daily to sustain remission of skin and muscle disease. In the 12 months since the dose was increased, our patient has not experienced any flares of disease.
留言 (0)