
記住我
Glioblastoma multiforme (GBM) is the most common primary brain tumor that affects over 17,000 people annually (1, 2). Despite extensive efforts to develop numerous novel therapies, GBM continues to produce poor outcomes, with an average 5-year survival of 6.9% and a mean survival of 15 months (3, 4). Even with standard-of-care therapies including surgical resection, temozolomide, and radiotherapy the average survival of patient’s only increased to 14.6 months (4, 5).
Classically, GBM pathogenesis involves a multitude of cellular adaptations and signaling cascades that cultivate a proliferative and immunosuppressive tumor microenvironment (6, 7). Therefore, altering the tumor immuno-microenvironment and targeting the immune system to attack and eliminate cancer cells is an attractive approach. Recently, chimeric antigen receptor T-cell therapy is a revolutionary form of cellular immunotherapy, which has produced remarkably effective results in hematological malignancies (8–14). In short, chimeric antigen receptors are lymphocytes engineered to recognize and eliminate cells expressing specific antigens. CAR’s are most commonly T cells which consist of four main components (a) an extracellular antigen binding domain, which recognizes the target antigen of interest, (b) a hinge region, which is the extracellular region that extends the antigen binding domain from the transmembrane domain, (c) the transmembrane domain, which functions as an anchor to the T cell membrane, and (d) one or more intracellular signaling domains (8). Figure 1 depicts the structure and mechanism of anti-EGFRVIII and EGFR CAR T cells.
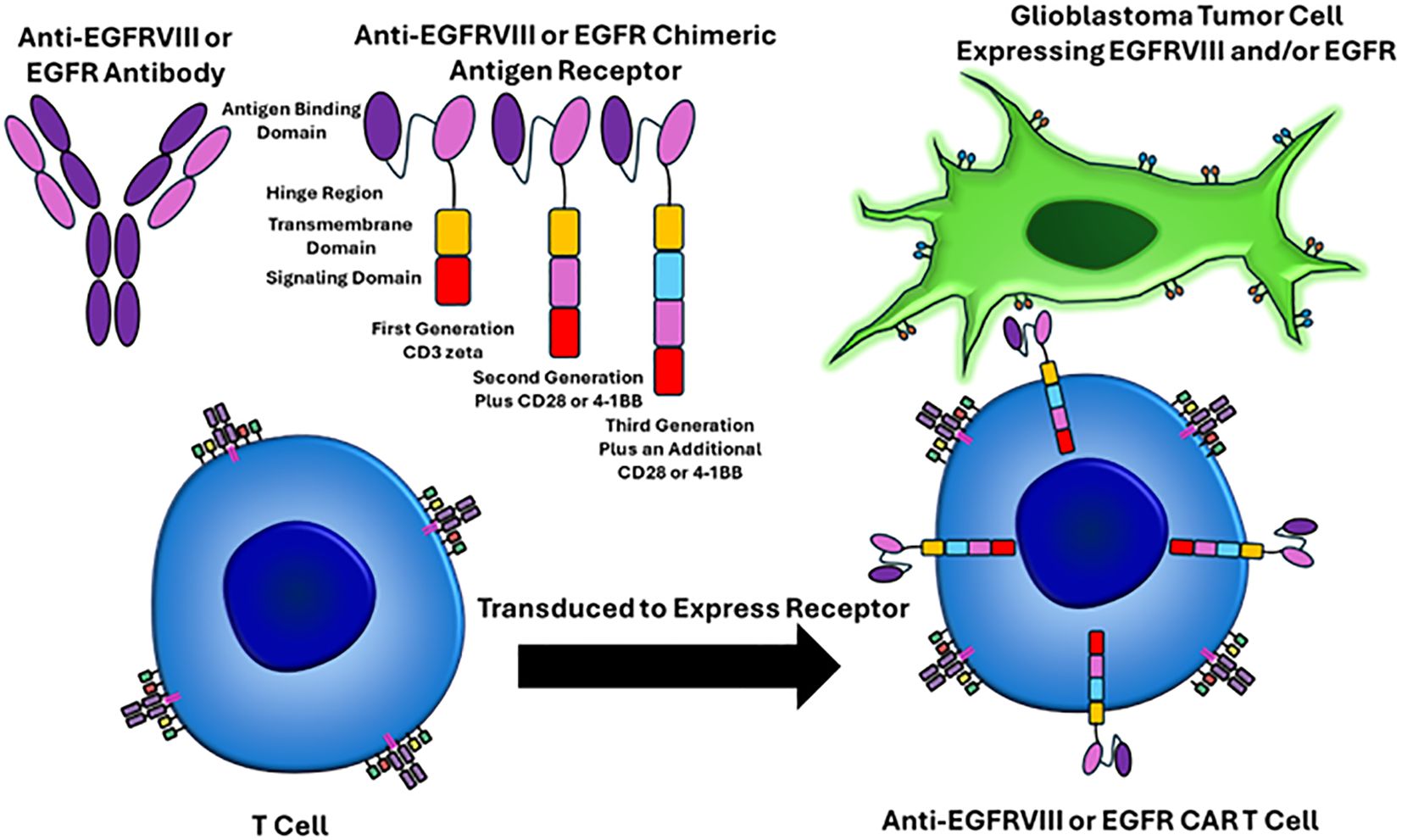
Figure 1. EGFRVIII and EGFR targeted CAR T cell mechanisms of action. The CAR T receptor targets EGFRVIII or EGFR on glioblastoma cells via single-chain variable fragments (antigen binding domain) that recognize epitopes on these proteins. The hinge region is the extracellular region that extends from the antigen binding domain to the transmembrane domain. The transmembrane domain functions as an anchor to the T cell membrane. One or more intracellular signaling domains are present. First generation CAR T cells only had a CD3 zeta chain and were not very effective. Second generation CAR T cells added an additional signaling domain, including CD28 or 4-1BB. Third generation CAR T cells have three intracellular signaling domains. T cells are harvested from the patient through apheresis, virally transduced to express the CAR T cell receptor, the CAR T cells are infused back into the patient, and then the CAR T cell can recognize the epitope of interest on the surface of the tumor cells.
In hematologic malignancies, for example, anti-CD19 CAR T cell therapy directed against B-cell malignancies was approved by the United States Food and Drug Administration in 2017 (11–13). Theoretically, this novel cancer treatment could be applied to solid tumors to potentially improve patient outcomes. Unfortunately, however, there are still several difficulties that must be addressed including limited efficacy against solid tumors due to multiple mechanisms, including but not limited to antigen escape, limited persistence, poor trafficking and tumor infiltration, and the immunosuppressive microenvironment (8, 9). In regard to GBM, multiple CAR T cell target antigens have been identified and CARs have been generated and tested in both preclinical and clinical studies (15, 16). One potentially exciting antigen is the epidermal growth factor receptor (EGFR) antigen, as it is frequently amplified in glioblastoma and a mutant variant is only expressed on the tumor (15–17). In this review, we will examine the preclinical and human study results of EGFR-targeted CAR T cell therapies and discuss current limitations and propose future directions.
2 EGFR epitopeEGFR is classically amplified and plays a key role in the progression and development of solid tumors including glioblastoma, non-small cell lung cancer, and breast, gastroesophageal, and colorectal cancer (18–20). In over 50% of GBMs, EGFR is amplified (17). Because of the high expression rate and role that EGFR plays in alteration of tumor development, progression, angiogenesis, invasion, and survival, EGFR is an exciting potential target for immunotherapy (4, 17).
3 EGFRVIII epitopeA common GBM associated variant of EGFR, Epidermal Growth Factor Receptor Variant III (EGFRVIII), is frequently expressed and correlates with the pathogenesis of GBM (15–17, 21, 22). More specifically, EGFRVIII is associated with tumor progression, invasiveness, and shorter survival (15–17, 21, 22). EGFRVIII is a result of an 801 base pair in-frame deletion from exons 2 to 7 (23–25).
4 EGFRVIII CAR T cells minimize on-target off-tumor toxicitiesImportantly, the variant structure of the extracellular domain EGFR VIII has been shown to be able to be targeted by monoclonal antibodies in a specific manner, which reduces the risk of systemic on-target, off-tumor toxicities (26–28). Because of the fact that EGFRVIII is only expressed on the GBM, it is a promising target as theoretically CAR T cells targeting EGFRVIII would avoid one of the major limitations of CAR T cell therapy, on-target-off-tumor effects (26–28). The benefit of targeting the CARs to EGFRVIII is that this antigen is only expressed in the tumor. This significantly lowers the risk of toxicity when using multiple CARs. Consideration of toxicities because of off-target effects is critical and must be assessed clinically to minimize the potential devastating effects of toxicities. Because of this, multiple strategies have been used to develop CAR T cell therapies and vaccines targeting EGFRVIII (26–29). In initial preclinical studies, EGFRVIII demonstrated significant killing of tumor cells (29). For example, Miao et al. demonstrated that the survival rate of mice was significantly enhanced, the EGFRVIII CAR T cells proliferated, and the growth of tumor cells was reduced in mice treated with EGFRVIII targeted CAR T cells (30). All of these preclinical studies were exciting as they demonstrated excellent killing ability (29, 30).
Studies in human subjects and randomized control trials, unfortunately, have produced more heterogeneous results. The first clinical study involving intravenous administration of autologous CAR T cells targeting EGFR VIII for recurrent glioblastoma occurred in 2017 (28). O’Rourke et al. reported the findings of the first 10 cases of recurrent glioblastoma treated with EGFR VIII-targeted CAR T cells. In this study, 7 patients underwent surgical intervention. Post-surgical analysis of the tissue showed that EGFRVIII targeted CARs trafficked to the tissue, and importantly, there was significant antigen reduction observed in 5 of the 7 patients (28). NCT02209376, showed anti-tumor effective CAR T cells with a median survival of 8 months in all patients (28). A novel single-chain fragment variable (scFv) that targets EGFRVIII, GCTO2, was constructed by Abbott et al. and injected to assess the efficacy of GCTO2 EGFRVIII-targeted CAR T cells using the U87 EGFRV3 glioblastoma model (31). After a single intravenous injection of GCTO2 CAR T cells, significant regression of tumor cells was demonstrated suggesting a very high specificity for EGFRV3 (31). Further evaluation, however, showed that the affinity of this CAR was significantly lower compared to clone 2173 scFv (31). Notably, many other CARs targeting other antigens on GBM including CAR T cells for GBM therapy are ephrin type-A receptor 2 (EphA2) (NCT02575261), and mucin 1 (MUC1) (NCT02839954, NCT02617134) are currently being developed. In addition, IL13Rα2-targeted CAR T cells are being worked on for glioblastoma. A patient with multifocal glioblastoma that received IL13Rα2 CAR T cells showed regression of all observed tumors for 7.5 months with no grade 3 or higher toxic effects (32). A phase 1 clinical trial in patients with recurrent high-grade glioma receiving IL13Rα2-targeted CAR T cells showed stable disease or better being seen in 50% of the treated patients and was safe (33). However, extensive review of these targets is beyond the scope of this review.
5 Minimal immunologic toxicities associated with EGFRVIII CAR T cellsThe most common adverse events seen in patients undergoing clinical trials with third generation CAR T cells targeting EGFR VIII were related to the lymphodepleting chemotherapy, which in patients treated with CAR T cell therapy is used before CAR T cell therapy infusion (28, 34, 35). Notably, 2 patients did have the side effect of severe dyspnea when CAR T cell therapy was administered at the highest dose with one of these patients ultimately dying (4, 28, 34). Several other patients had new minor changes in their neurological exam or seizures, which required treatment with antileptic medications or steroids (34). Importantly, however, no patients ended up developing significant side effects from an immunological standpoint, which historically has been a severely limiting side effect in hematologic malignancies (28). Although toxicities are less of a concern for EGFRVIII target therapies because of the high specificity of the antigen for the tumor cells itself, toxicities for CAR T cell therapy are an important limitation of CAR T cells in general and are a critical factor in the design of the CAR. Design of the antigen binding domain, hinge region, transmembrane domain, and signaling domains all can be optimized to potentially reduce toxicities and careful selection of dual antigens will be necessary to not only improve therapy but also continue to keep toxicity low.
6 EGFRVIII CAR T cells and the tumor microenvironmentAlthough there is exciting anecdotal data, including a patient with a history of recurrent GBM who received EGFRVIII targeted CAR T-cell therapy who ended up surviving for 36 months after treatment with CAR T cell therapy, these responses have not been repeatably observed in other patients (35). For instance, a pilot trial demonstrated that anti-EGFRVIII CAR T cells showed no significant clinical efficacy in treating GBM in a phase I pilot trial (34). Unfortunately, although EGFRVIII targeted CARs can be a useful target as they are relatively highly expressed and limit on-target off-tumor toxicities, the progression-free survival in some studies has only been a month while the overall median survival after treatment is only 6.9 months (34). Therefore, it is critical to understand that in order to get the most robust efficacy from EGFR targeted CAR T cells, the tumor microenvironment must also be targeted and manipulated. The immunosuppressive microenvironment is a key consideration as many cell types that drive immunosuppression including myeloid derived suppressor cells, tumor associated macrophages, and regulatory T cells can infiltrate solid tumors and drastically reduce the effectiveness of CAR T cell therapy as these cells produce tumor facilitating cytokines, chemokines, and growth factors. Strategies to ameliorate this immunosuppressive microenvironment include the utilization of immune checkpoint inhibitors with CAR T cell therapy. This investigation of immune checkpoint inhibitors with CAR T cell therapy is ongoing multiple types of solid tumors including gliomas. Combination immune checkpoint inhibitor and CAR T cell therapy has produced excellent results hematological malignancies with great potential in solid tumors. Although theoretically combining CAR T cell therapy targeting EGFRVIII with immune checkpoint therapy should significantly improve efficacy, a recent Phase I trial unfortunately demonstrated no efficacy in these patients, suggesting the need for additional strategies (36). In a preclinical study, metformin and rapamycin pre-treatment of EGFRvIII CAR-T cells improved metabolic states of the CAR T cells in the hypoxic microenvironment of glioblastoma and prolonged mouse survival (37). Other combination therapy strategies that have been attempted with CAR-T cells mainly in hematological malignancies and in some solid tumors include immunomodulators, BiTEs, vaccines, radiation, oncolytic virus, dendritic cell vaccines, cytokine or cytokine receptor blocker therapy, BTK inhibitor, PI3-kinase inhibitor, LAG3 inhibitor, dimerizer drug, stem cell transplantation, chemotherapy, and corticosteroids (38). Specifically, with CAR T cell combinations in solid tumors, strategies have included trying to increase tumor infiltration and tumor recognition with DDR inhibitors, angiogenesis inhibitors, MAPK inhibitors, and EZH2 inhibitors; trying to enhance cytotoxicity and reduce exhaustion with lenalidomide, TLR agonists, metabolic regulators, BET inhibitors, and DNMT inhibitors; and trying to increase memory phenotype with TET2 inhibitors, HDAC inhibitors, metabolic regulators, PI3K-AKT inhibitors, LCK/FYN inhibitors, and JAK inhibitors (39). Further study is necessary to tune the microenvironment to be more responsive to CAR T cell therapy. Efforts have also focused on engineering CARs that are resistant to immunosuppressive factors in the hostile tumor microenvironment such as TGF-beta (40). Many studies are also looking at how to create CARs with numerous cytokines to create armored CARs, which express pro-inflammatory cytokines instead of just focusing on inhibitory signals. Thus, further study is necessary to tune the microenvironment to be more responsive to CAR T cell therapy.
The tumor microenvironment presents a significant challenge to the field as it not only serves as a physical barrier but also is immunosuppressive. Thus, the tumor microenvironment prevents CAR T cells from properly trafficking and infiltrating solid tumors drastically reducing their efficacy. In order to obtain the efficacy seen in hematological malignancies, it will likely require multiple technical advances to circumvent the immunosuppressive microenvironment. Multiple approaches are currently being investigated to improve trafficking and infiltration. One such method involves the utilization of delivery routes other than systemic delivery. Currently, intraventricular injection and other local administration routes versus systemic delivery are currently under investigation (41). It is important to note that local delivery, including intraventricular administration, does not require lymphodepletion, thus avoiding the side effects discussed above. Additionally, local delivery allows for repeated administration, which is advantageous. Another exciting approach is to design CARs that express chemokine receptors that match and respond to tumor derived chemokines. These chemokine receptor expressing CARs have been shown to be possibly effective in CAR T cell therapies targeting other solid tumors and further investigation in glioblastoma is needed (42). Another strategy takes advantage of the fact that the tumor stroma extracellular matrix is composed of heparin sulfate proteoglycan (43). CAR T cell therapies can be engineered to express heparinases which function to degrade this physical barrier. These heparinase expressing CARs are currently under investigation and application to GBM is worth investigation. One possible approach to limit the immunosuppressive tumor microenvironment is to inhibit vascular endothelial growth factor, as inhibition of VEGF not only normalizes tumor vessels in both human and mouse models, but also improves the delivery of CD8 positive T cells and the effectiveness of immunotherapies. Excitingly, a recent study found that treatment with anti-mouse VEGF antibodies improved both the distribution and infiltration of CAR T cells targeting EGFR VIII in GBM, which resulted in both prolonged survival and slowed tumor growth compared to EGFR VIII CAR T cell therapy by itself (44).
7 EGFRVIII CAR T cells and antigen escapeAs with many solid tumors, targeting one antigen using CAR T cell therapy is currently insufficient to produce the drastic responses seen in hematological malignancies. The current limitations of CAR T cell therapy’s efficacy against solid tumors including glioblastoma are due to multiple mechanisms of tumor resistance to single-targeted CAR T cell construct. One important consideration is the mechanism of antigen escape. Although initially tumor cells may express high levels of an antigen, CAR T cell therapies targeting these cells will result in either partial or complete loss of the target antigen expression as the CAR eliminates these cells from the tumor. In other words, the CAR is naturally selecting tumor cells without the antigen by killing those with the antigen. A strategy to circumvent this limitation is to use dual-targeted CAR T cell therapy in which chimeric antigen receptors targeting two or more antigens are use. These antigens can be targeted by using two CAR T cells that each target a single antigen or through the use of tandem CARs, which is a single CAR construct that contains two single-chain variable fragment antigen recognition domains in order to concomitantly target multiple target tumor antigens (45–47). Tandem CAR T-cells targeting EGFRVIII and IL-13R α2 appear to be a potentially attractive approach based off preclinical data using patient-derived xenograft mouse models, which demonstrated long term durable and complete responses against heterogeneous GBM tumors (48). A syn Notch receptor that recognizes a specific priming antigen, such as the heterogeneous but tumor-specific glioblastoma neoantigen epidermal growth factor receptor splice variant III (EGFRvIII) or the central nervous system (CNS) tissue-specific antigen myelin oligodendrocyte glycoprotein (MOG), locally induces expression of a CAR (49). This allows for killing by targeting antigens that are homogeneous but not absolutely tumor specific and reduces exhaustion (49). EGFRvIII synNotch-CAR T cells showed higher antitumor efficacy and T cell durability than their regular CAR T cell counterparts without off-tumor effects (49). T cells with a synNotch-CAR circuit primed by MOG also resulted in good control without priming outside the brain (49).
8 EGFR CAR T cells for glioblastomaAn unintended consequence of EGFRVIII-targeted CARs, however, is that it can lead to the proliferation of EGFRVIII-negative-EGFR-positive glioblastoma, an example of antigen escape in action (50). An exciting potential solution to this limitation is the creation of CAR T bispecific T cell engager (BiTE) cells (50). These CAR-T BiTE cells have the ability to secrete EGFR-specific BiTEs, which have shown the ability to eliminate heterogeneous tumors in glioblastoma mouse models through recruiting untransduced T cells against wild-type EGFR and redirecting CAR T cells (50). Excitingly, this approach also appears to not produce the toxicity seen with EGFR-targeted CAR T cells (50). In a first in human study, three recurrent glioblastoma patients were treated with CARv3-TEAM-E T cells, which are chimeric antigen receptor (CAR) T cells engineered to target the epidermal growth factor receptor (EGFR) variant III tumor-specific antigen, as well as the wild-type EGFR protein, through secretion of a T-cell-engaging antibody molecule (TEAM) (51). The treatment appeared to be safe. There was regression of the tumor, but this was transient in 2 of 3 patients (51). Another preclinical model involving human-induced pluripotent stem cell-derived teratoma xenograft model showed that EGFR806 CAR T cells, which are able to recognize low density EGFR, both selectively and effectively targeted EGFR-expressing tumor cells (52). Similarly, Chen et al. showed that an EGFR806 targeting CAR was effective in vitro and in vivo in a dose dependent manner (29).
Excitingly, interim results of a Phase I clinical trial assessing intrathecal efficacy of bivalent CAR-T cells targeting EGFR and IL-13R-alpha-2 in six recurrent glioblastoma patients demonstrated improvements in tumor size and enhancement, substantial CAR T cell abundance, and significant cytokine release in the cerebrospinal fluid in all six patients (53). These results are promising; however, longer follow-up time and a higher sample size are needed for more definitive conclusions. HER2 also appears to be an effective co-target in glioblastoma (54, 55). Other studies have used CARs targeting HER2 and IL-13RA2 in glioblastoma and produced some initial promising results (56). Further studies using EGFR-directed CAR T cells in combination with other targeted antigens are warranted in the future.
A potential limitation of using EGFR-targeted CAR T cells is on-target, off-tumor effects as EGFR is more widely expressed throughout the body than the more specific EGFVIII and thus a higher chance of CAR T associated toxicities ensues. The tumor microenvironment and antigen escape remain challenges with EGFR as a therapeutic target as well.
9 DiscussionEGFRVIII and EGFR as CAR T cell targets are exciting as they are commonly amplified in glioblastoma. A summary of the studies covered in this review are shown in Table 1. Because of the fact that EGFRVIII is only expressed on the GBM, it is a promising target as theoretically CAR T cells targeting EGFRVIII would avoid one of the major limitations of CAR T cell therapy, on-target-off-tumor effects (26–28). A potential limitation of using EGFR-targeted CAR T cells is on-target, off-tumor effects as EGFR is more widely expressed throughout the body than the more specific EGFVIII and thus a higher chance of CAR T associated toxicities ensues. The EGFRVIII has theoretical potential to be a fantastic and an efficacious target as it is only expressed on tumor cells. Although pre-clinical studies demonstrate remarkable killing, human studies unfortunately have produced heterogeneous results that suggest further optimization and strategies are needed in order to continue to push CAR T cell therapy towards improving patient outcomes. The tumor microenvironment presents a significant challenge to the field as it not only serves as a physical barrier but also is immunosuppressive. Thus, the tumor microenvironment prevents CAR T cells from properly trafficking and infiltrating solid tumors drastically reducing their efficacy. The immunosuppressive microenvironment is a key consideration as many cell types that drive immunosuppression including myeloid derived suppressor cells, tumor associated macrophages, and regulatory T cells can infiltrate solid tumors and drastically reduce the effectiveness of CAR T cell therapy as these cells produce tumor facilitating cytokines, chemokines, and growth factors. Although theoretically combining CAR T cell therapy targeting EGFRVIII with immune checkpoint therapy should significantly improve efficacy, a recent Phase I trial unfortunately demonstrated no efficacy in these patients, suggesting the need for additional strategies (36). As with many solid tumors, targeting one antigen using CAR T cell therapy, including targeting EGFRVIII or EGFR, is currently insufficient to produce the drastic responses seen in hematological malignancies due to antigen escape. Thus, while targeting EGFRVIII or EGFR in glioblastoma via CAR T cell therapy holds promise, there are still many obstacles to be overcome before these strategies can become effect clinical treatments.
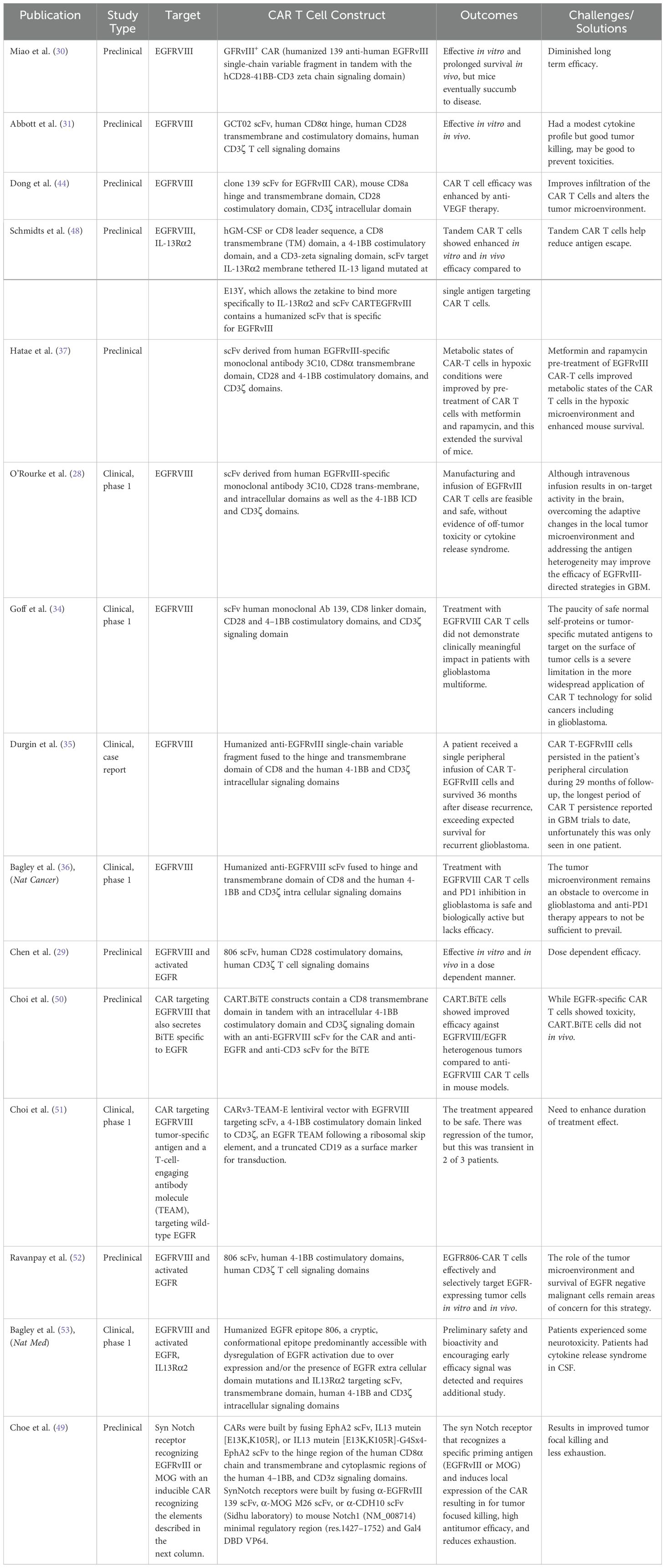
Table 1. Summary of EGFRVIII and EGFR CAR T cell studies in glioblastoma.
Author contributionsRCS: Conceptualization, Writing – original draft, Writing – review & editing. RMS: Conceptualization, Supervision, Writing – original draft, Writing – review & editing.
FundingThe author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interestRMS is an inventor on patents related to CAR T cell therapy licensed to Humanigen through Mayo Clinic.
The remaining author declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References1. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncology. (2021) 23:1231. doi: 10.1093/neuonc/noab106
PubMed Abstract | Crossref Full Text | Google Scholar
2. Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, et al. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide on research studies in the United States. Clin Cancer research. (2010) 16:2443. doi: 10.1158/1078-0432.CCR-09-3106
Crossref Full Text | Google Scholar
4. Luksik AS, Yazigi E, Shah P, Jackson CM. CAR T cell therapy in glioblastoma: overcoming challenges related to antigen expression. Cancers (Basel). (2023) 15:1414. doi: 10.3390/cancers15051414
PubMed Abstract | Crossref Full Text | Google Scholar
5. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
PubMed Abstract | Crossref Full Text | Google Scholar
6. Esemen Y, Awan M, Parwez R, Baig A, Rahman S, Masala I, et al. Molecular pathogenesis of glioblastoma in adults and future perspectives: A systematic review. Int J Mol Sci. (2022) 23:2607. doi: 10.3390/ijms23052607
PubMed Abstract | Crossref Full Text | Google Scholar
7. Rajarathnam K, Rösgen J. Isothermal titration calorimetry of membrane proteins — Progress and challenges. Biochim Biophys Acta (BBA) - Biomembranes. (2014) 1838:69–77. doi: 10.1016/j.bbamem.2013.05.023
Crossref Full Text | Google Scholar
9. Sterner RC, Sterner RM. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front Immunol. (2022) 13:879608. doi: 10.3389/fimmu.2022.879608
PubMed Abstract | Crossref Full Text | Google Scholar
10. Sterner R, Kenderian S. Myeloid cell and cytokine interactions with chimeric antigen receptor-T-cell therapy: implication for future therapies. Curr Opin Hematol. (2020) 27:41–8. doi: 10.1097/MOH.0000000000000559
PubMed Abstract | Crossref Full Text | Google Scholar
12. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
PubMed Abstract | Crossref Full Text | Google Scholar
13. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
PubMed Abstract | Crossref Full Text | Google Scholar
15. Congdon KL, Gedeon PC, Suryadevara CM, Caruso HG, Cooper LJN, Heimberger AB, et al. Epidermal growth factor receptor and variant III targeted immunotherapy. Neuro Oncol. (2014) 16 Suppl 8:viii20–25. doi: 10.1093/neuonc/nou236
PubMed Abstract | Crossref Full Text | Google Scholar
16. Eskilsson E, Rosland GV, Talasila KM, Knappskog S, Keunen O, Sottoriva A, et al. EGFRvIII mutations can emerge as late and heterogenous events in glioblastoma development and promote angiogenesis through Src activation. Neuro Oncol. (2016) 18:1644–55. doi: 10.1093/neuonc/now113
PubMed Abstract | Crossref Full Text | Google Scholar
17. Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res. (2005) 11:1462–6. doi: 10.1158/1078-0432.CCR-04-1737
PubMed Abstract | Crossref Full Text | Google Scholar
19. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci. (2016) 59:468–79. doi: 10.1007/s11427-016-5023-8
PubMed Abstract | Crossref Full Text | Google Scholar
20. Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Res. (2000) 60:3081–7.
PubMed Abstract | Google Scholar
22. Chistiakov DA, Chekhonin IV, Chekhonin VP. The EGFR variant III mutant as a target for immunotherapy of glioblastoma multiforme. Eur J Pharmacol. (2017) 810:70–82. doi: 10.1016/j.ejphar.2017.05.064
PubMed Abstract | Crossref Full Text | Google Scholar
23. Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. (1997) 57:4130–40.
PubMed Abstract | Google Scholar
27. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. (2017) 18:1373–85. doi: 10.1016/S1470-2045(17)30517-X
PubMed Abstract | Crossref Full Text | Google Scholar
28. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9. doi: 10.1126/scitranslmed.aaa0984
PubMed Abstract | Crossref Full Text | Google Scholar
29. Chen M, Sun R, Shi B, Wang Y, Di S, Luo H, et al. Antitumor efficacy of chimeric antigen receptor T cells against EGFRvIII-expressing glioblastoma in C57BL/6 mice. BioMed Pharmacother. (2019) 113:108734. doi: 10.1016/j.biopha.2019.108734
PubMed Abstract | Crossref Full Text | Google Scholar
30. Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, Leon GD, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PloS One. (2014) 9:e94281. doi: 10.1371/journal.pone.0094281
PubMed Abstract | Crossref Full Text | Google Scholar
31. Abbott RC, Verdon DJ, Gracey FM, Hughes-Parry HE, Iliopoulos M, Watson KA, et al. Novel high-affinity EGFRvIII-specific chimeric antigen receptor T cells effectively eliminate human glioblastoma. Clin Transl Immunol. (2021) 10:e1283. doi: 10.1002/cti2.1283
Crossref Full Text | Google Scholar
32. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
PubMed Abstract | Crossref Full Text | Google Scholar
33. Brown CE, Hibbard JC, Alizadeh D, Blanchard MS, Natri HM, Wang D, et al. Locoregional delivery of IL-13Rα2-targeting CAR-T cells in recurrent high-grade glioma: a phase 1 trial. Nat Med. (2024) 30:1001–12. doi: 10.1038/s41591-024-02875-1
PubMed Abstract | Crossref Full Text | Google Scholar
34. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor transduced T cells targeting EGFRvIII in patients with glioblastoma. J immunotherapy (Hagerstown Md : 1997). (2019) 42:126. doi: 10.1097/CJI.0000000000000260
Crossref Full Text | Google Scholar
35. Durgin JS, Henderson F, Nasrallah MP, Mohan S, Wang S, Lacey SF, et al. Case report: prolonged survival following EGFRvIII CAR T cell treatment for recurrent glioblastoma. Front Oncol. (2021) 11:669071. doi: 10.3389/fonc.2021.669071
PubMed Abstract | Crossref Full Text | Google Scholar
36. Bagley SJ, Binder ZA, Lamrani L, Marinari E, Desai AS, Nasrallah MP, et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat Cancer. (2024) 5:517–31. doi: 10.1038/s43018-023-00709-6
PubMed Abstract | Crossref Full Text | Google Scholar
37. Hatae R, Kyewalabye K, Yamamichi A, Chen T, Phyu S, Chuntova P, et al. Enhancing CAR-T cell metabolism to overcome hypoxic conditions in the brain tumor microenvironment. JCI Insight. (2024) 9. doi: 10.1172/jci.insight.177141
PubMed Abstract | Crossref Full Text | Google Scholar
38. Al-Haideri M, Tondok SB, Safa SH, Maleki AH, Rostami S, Jalil AT, et al. CAR-T cell combination therapy: the next revolution in cancer treatment. Cancer Cell Int. (2022) 22:365. doi: 10.1186/s12935-022-02778-6
PubMed Abstract | Crossref Full Text | Google Scholar
39. Liu J, Jiao X, Ma D, Fang Y, Gao Q. CAR-T therapy and targeted treatments: Emerging combination strategies in solid tumors. Med. (2024) 5:530–49. doi: 10.1016/j.medj.2024.03.001
PubMed Abstract | Crossref Full Text | Google Scholar
40. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-β Receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
PubMed Abstract | Crossref Full Text | Google Scholar
41. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2+ Breast cancer metastasis to the brain. Clin Cancer Res. (2018) 24:95–105. doi: 10.1158/1078-0432.CCR-17-2041
PubMed Abstract | Crossref Full Text | Google Scholar
42. Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-cells targeting the integrin αvβ6 and co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid Malignancies. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11050674
PubMed Abstract | Crossref Full Text | Google Scholar
43. Zhang B-L, Qin D-Y, Mo Z-M, Li Y, Wei W, Wang Y-S, et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci. (2016) 59:340–8. doi: 10.1007/s11427-016-5027-4
PubMed Abstract | Crossref Full Text | Google Scholar
44. Dong X, Ren J, Amoozgar Z, Lee S, Datta M, Roberge S, et al. Anti-VEGF therapy improves EGFR-vIII-CAR-T cell delivery and efficacy in syngeneic glioblastoma models in mice. J Immunother Cancer. (2023) 11:e005583. doi: 10.1136/jitc-2022-005583
PubMed Abstract | Crossref Full Text | Google Scholar
45. Zhang H, Gao L, Liu L, Wang J, Wang S, Gao L, et al. A Bcma and CD19 bispecific CAR-T for relapsed and refractory multiple myeloma. Blood. (2019) 134:3147–7. doi: 10.1182/blood-2019-131056
留言 (0)