
記住我








Two-group randomized controlled trial (RCT) of active versus sham rTMS. rTMS uses pulses of magnetic energy to non-invasively stimulate the brain. The rTMS device to be used in this study is the Magstim Rapid2. The rTMS device can either connect to a “active” coil (used in the active group) which rests against the scalp and delivers the magnetic energy to the brain or can be connected to an identically appearing “sham” coil (used in the sham group) that also rests against the scalp but does not deliver any magnetic energy to the brain. The rTMS protocol is specified by the parameters of pulse frequency, duration of pulse delivery (“train duration”), intensity, and stimulation location.
Our study proposes to have the active rTMS group receive a previously described rTMS protocol for treatment of patients with chronic pain in research studies (a “high-frequency protocol”) [4]: a pulse frequency of 10 pulses per second, with 10-s pulse trains, at an intensity of 80% of resting motor threshold, delivered over the pelvic-SMA region, defined in Montreal Neurological Institute (MNI) coordinates to be X = − 2, Y = − 16, and Z = 68 mm. A “session” of rTMS in our study will consist of 20 pulse trains, each separated by 50 s. Should protocol changes be needed, the proposed changes are submitted to the DSMB first, then the home institute’s internal review board (IRB) to be reviewed and approved. Protocol changes may not be implemented until approved. Any protocol deviations are reported to the IRB to be documented and acknowledged.
Study populationOur study population is actively being recruited through various avenues, including the University of Southern California’s broad medical system, focusing on, but not limited to those in the greater Los Angeles area. Additionally, recruitment has been mindful of inclusivity and equity within our system, recruiting from various settings, including but not limited to our USC Keck hospitals and LA County General Hospital, and outpatient PT clinics in the greater Los Angeles area, in hopes to improve diversity in our participants.
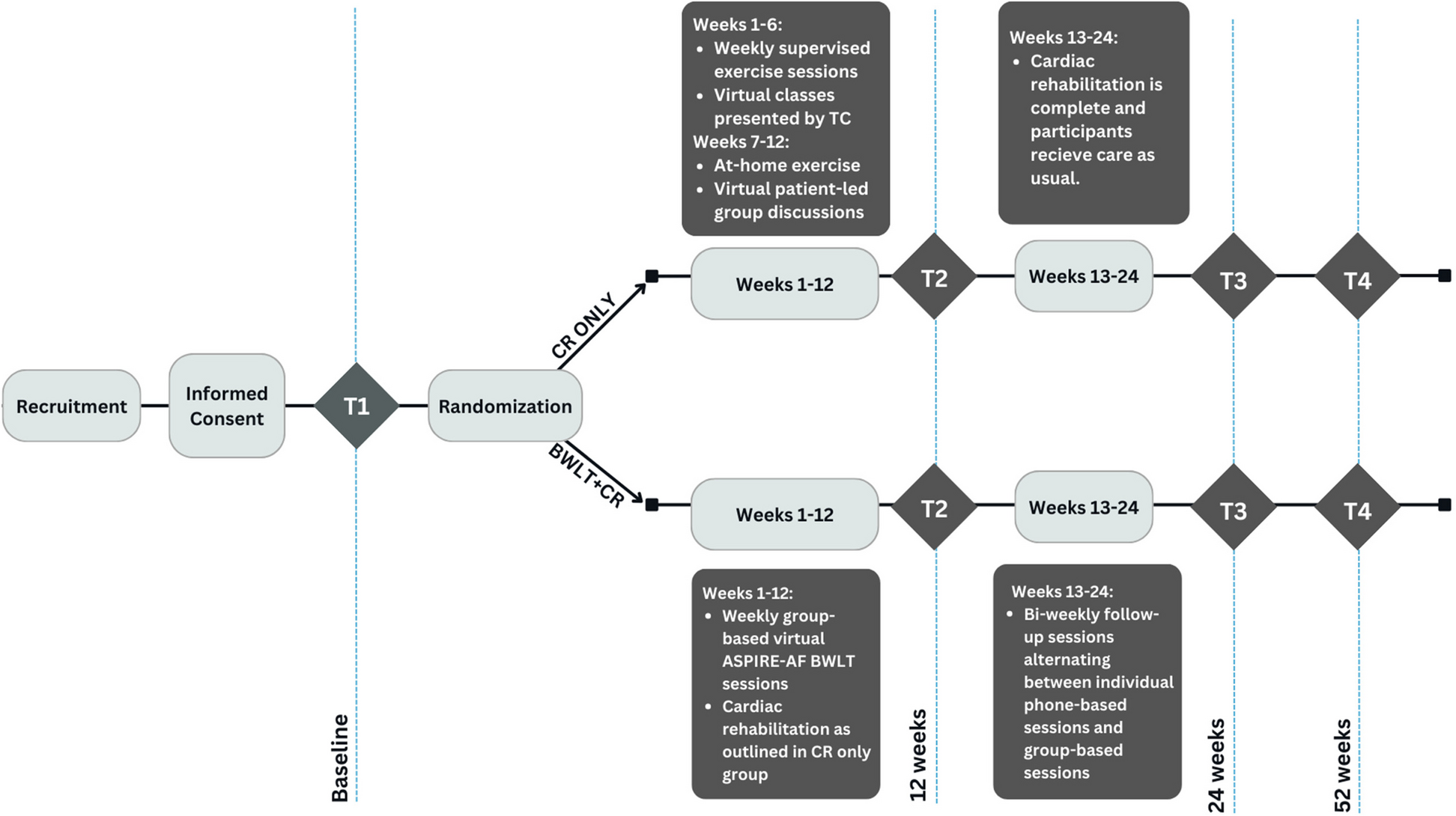
Study settingParticipants will be treated at the University of Southern California Division of Biokinesiology and Physical Therapy Applied Movement and Pain Laboratory (AMPL) with MRI being performed onsite at the USC Stevens Institute for Neuroimaging and Neuroinformatics. Study workflow is summarized in Fig. 1.
Fig. 1 Inclusion criteria
Inclusion criteria
Female at least 18 years old.
Have a diagnosis of IC/BPS by a physician.
Urologic symptoms present a majority of the time during the most recent 3 months.
Report 4/10 pain or greater pain in response to “At its worst, on scale 0–10 how bad did your pelvic pain get in the past 3 months?” asked at screening.
Exclusion criteriaThis study uses several measures to exclude individuals who have a known physical cause of their symptoms such as an active infection, urethral strictures, active cancer treatments/pelvic malignancies, and neurological conditions affecting the bladder. In addition, rTMS and fMRI pose higher than average health consequences to individuals with personal factors such as pregnancy, seizure disorders, and chronic headaches. As such, participants with these factors were designated ineligible for this study.
Participants will be excluded from the study if they report:
Symptomatic urethral stricture
On-going neurological conditions affecting the bladder or bowel
Active auto-immune or infectious disorders
History of cystitis caused by tuberculosis or radiation or chemotherapies
History of non-dermatologic cancer
Current major psychiatric disorders
Severe cardiac, pulmonary, renal, or hepatic disease
Conditions or the use of medical devices that are contraindications for either fMRI or rTMS procedures, including pregnancy, seizure disorders, or chronic headaches
Physicians with expertise in clinical TMS or IC/BPS are consulted if there are any questions about eligibility for the study, and their expert opinion documented.
Deferral criteria and managementParticipants are deferred if:
They test positive on the McKesson urinalysis test during the baseline visit.
They report having started any new treatments or medications for IC/BPS in the past 3 months during screening.
For either of the following, interested participants will be re-evaluated in no sooner than 6 weeks.
RecruitmentRecruitment for this study was conducted through the Applied Movement and Pain Laboratory (AMPL) in the Department of Biokinesiology and Physical Therapy at the University of Southern California (USC). This study worked with providers in the USC Medical System and outpatient Pelvic Health Physical Therapy centers in the greater Los Angeles area to identify potentially appropriate participants. Additionally, research coordinators used digital platforms such as YouTube, social media, and email to inform the broader population of this study as well as subscribers and members of the national support groups. Interested participants were then screened for appropriateness and safety for the trial.
Randomization, allocation concealment, and blindingWe will randomize treatment assignments using a randomized block sequence as implemented in the blockrand function in the R statistical package [5]. To ensure approximately equal allocation to the two arms throughout the study, randomization is conducted in blocks, of sizes 2, 4 and 6. The randomization order will be administered by a statistician independent of the participant recruitment and evaluation. A list of 50 record IDs was generated paired with a 4-letter code for the active and sham coil. As participants are enrolled in the study, they are assigned a record ID and matched to the subsequent coil/treatment group. Study staff delivering the intervention know the coil only by code and are specifically instructed that they must remain blinded to which coil is active. This is a double quadruple-blinded study where both the interventionists and participants are blinded to which treatment group they have been assigned (sham vs. active), as are the investigators and outcome assessors.
InterventionThe active and sham groups will undergo identical protocol, with the only difference being that the active vs. sham coil used per designated group for the 5-day treatment week. During the participant’s 6-week participation and treatment period, they will be asked to discontinue any other IC/BPS treatments and therapies, to reduce any confounding treatment variables. If this is not medically advised by the study physician, participants will be asked to report other concurrent treatments.
Outcome assessmentOutcomes will be assessed at the initial baseline visit, throughout the treatment week, and at a follow-up visit 3 weeks later. At baseline, participants will complete a urine dip test, subjective questionnaires, and a pelvic floor muscle exam. Day 1 of treatment will involve outcome assessment via subjective questionnaires, and fMRI pre- and post-rTMS, and pelvic floor muscle assessment during rTMS via a surface electromyographic (EMG) sensor that rests just inside the rectum. Treatments 2 through 4 involve outcome assessment via questionnaires and rTMS treatments. Treatment 5 involve outcome assessment via questionnaires (listed below) and rTMS treatment with pelvic floor muscle assessment. A follow-up visit 3 weeks later will include outcome assessment via questionnaires, fMRI, and pelvic exam.
Outcome measures assessed include demographic information, Genitourinary Pain Index (GUPI) [6], Interstitial Cystitis Symptom Index (ICSI) [7], Overactive Bladder Questionnaire (OAB-q) [8], Urinary Distress Inventory (UDI-6) [9], Complex Multisystem Inventory (CMSI) [10], Collaborative Health Outcomes Registry Body Map (CHOIR) [11], Patient-Reported Outcomes Measurement Information System (PROMIS) [12], Coping Strategies Questionnaire (CSQ) [13], Hospital Anxiety and Depression Scale (HADS) [14], and The Positive and Negative Affect Schedule (PANAS) [15].
Primary outcomesSubjective ratings of pain symptoms are the primary outcome measures of the study. Pain will be assessed using a visual analog scale (VAS): the scale ranges from 0 (no pain) to 10 (worst pain imaginable). Pain will also be assessed using a global response assessment (GRA) asking “As compared to when you started the study treatment, how would you rate your interstitial cystitis/bladder pain syndrome (IC/BPS) symptoms now?.” The 3 pain primary outcome measures are:
Longer-term VAS change: before first treatment to 3 weeks after last treatment
Shorter-term VAS change: before first treatment to 1 day later just before second treatment
GRA: 3 weeks after last treatment
Secondary outcomesSecondary outcomes from fMRI and pelvic EMG will be computed exactly as described previously [3]. In summary:
Change in fractional amplitude of low frequency fluctuations (fALFF) in pelvic-SMA determined from fMRI. This change will be assessed between 1 h before and one hour after first treatment.
Change in pelvic floor muscle activity from pelvic EMG. This change will be assessed between just before to between 5 and 10 min after the start of first treatment.
Safety and participant compensationTransient headache is one of the known side effects of rTMS treatment. During and following every TMS session, we will ask the participant about any incidence of headache and neck pain. If the participant complains of any discomfort or pain during the TMS session, the session will be terminated immediately. The participant’s symptoms and side effects will be monitored both during treatment and longer-term side effects will be assessed at the follow-up visit (Fig. 2). There is no anticipated harm and compensation for trial participation and there are no plans to offer any type of payment for injury. If the participant requires treatment because they were injured, from participating in the study, treatment will be provided and the participants’ health insurance plan will be billed for treatment. The study sponsor will not pay for this treatment. The rTMS is being performed at a low intensity (80% of the FDI-thresholding) and the treatment duration is brief; no dosage changes are proposed. If a participant were to miss a treatment session, they can continue with the remaining scheduled visits and no treatment sessions will be rescheduled. Any moderate or severe side effect will be reported to the IRB as an adverse event within 24 h.
Fig. 2
Study schedule of enrolment, event, and assessments
The participant can withdraw from the study at any time for any reason. Treatment will be discontinued and all side effects will be documented. The study personnel will notify the PI as soon as any possible adverse event occurs and the PI will submit the adverse event reports. IRB will be notified immediately of all adverse and serious adverse events and the DSMB will be notified, as well as receive an annual summary of all adverse events.
Oversight and monitoringA 3-member Data and Safety Monitoring Board (DSMB) has been established for this study in accordance with NIH guidelines. The study’s full case report forms and standard operating procedures (CRF + SOP) stored in secure files. All data collected will be stored in a secure REDCap database without any participant identifying information. Identifying information will be kept securely by the principal investigator (PI). A single password-protected spreadsheet will be maintained by the PI with the participant’s name and contact information as a key to codes and this spreadsheet will be destroyed at study completion to ensure all data analyses are de-identified. We will do an annual progress report, continued review and DSMB meeting to go over the status of the study and compliance. Should any other concern for patient safety occur throughout the study, the DSMB is consulted immediately.
Sample sizeWe designed our study to have N = 50 participants, with N = 25 for each of the two groups (N = 25 sham, N = 25 high-frequency rTMS), to provide 84% power for the primary hypothesis. The primary hypothesis will be tested with a two-sample t-test for a difference in mean pain outcome measure in the high-frequency rTMS group versus the sham group. We performed this power calculation using G*Power version 3.1 under the assumptions of a two-sided hypothesis test, a type I error rate of 0.05, and the following effect sizes from the literature. In a prior study with N = 13 [16], pain (measured on a 100-point scale) was reduced an average of 15 points in the high-frequency rTMS group (SD = 20) and 3 points in the sham group (SD = 22) at 3 weeks after the last rTMS session, compared to baseline.
Statistical analysesData will be analyzed using R [17] by a blinded statistician not involved with participants or data collection. Initiative descriptive analyses will include evaluation of the distributions of all variables to evaluate modeling assumptions (e.g., Shapiro–Wilk test for normality) and identify potential extreme values. All data will be included in the primary analysis, but any extreme values will be discussed with the study team and potentially excluded in a sensitivity analysis should the extreme value be suspected to be erroneous. Hypothesis tests will be two-tailed, using a significance level of α = 0.05. The primary hypotheses will be evaluated using a two-sample t-test for a difference in mean pain outcome between the two groups, assuming normality of the outcome (or its natural log transformation) in each group. If normality assumptions are not satisfied, we will instead use the nonparametric alternative to the two-sample t-test, the Wilcoxon rank sum test to test for a difference in medians between the two groups. The number of adverse events will be tabulated and compared across the two groups using a chi-square test or Fisher’s exact test. Final primary hypothesis results and R code for producing these results will be presented to the Independent Monitoring Committee by the blinded statistician for certification. The independent DSMB statistician will unblind the results and assign the true group labels. Secondary and tertiary analyses will be performed unblinded, using two-sample t-tests (or Wilcoxon rank sum tests) and standard mediation analysis methods [18].
Collection and management of dataParticipants will receive seven in-person visits at the University of Southern California Health Science Campus Applied Movement and Pain Laboratory (AMPL) over the course of approximately 1 month. On the first (baseline) in-person visit, the participant will be tested to rule out any urinary tract infection (UTI) and pending the test is negative, they will proceed with the process of collecting EMG data and a pelvic examination. Additionally, the participant will complete a series of online questionnaires. Treatment week, which consists of 5 in-person (Monday–Friday) sessions, is then scheduled no greater than 2 weeks post-baseline visit. In the 3 days preceding this treatment week, the participants will fill out several online questionnaires pertaining to their pain level each morning, as well as the same questionnaires prior to each rTMS treatment. During the treatment week, the participant will receive rTMS treatment, based on their randomization group (treatment vs. sham). On the first of the 5-day treatments, brain imaging fMRI will be taken before and after brain stimulation (rTMS). Additionally, the use of a rectal EMG sensor will be used Monday and Friday during rTMS treatment.
The final portion of data collection occurs 3 weeks post Friday of the 5-day treatment week, preceded by 3 days of the same questionnaires to understand any changes in their pelvic pain or urinary urgency. During this final in-person visit, the participant completed the same set of questionnaires administered at the baseline visit, followed by fMRI, an EEG, and pelvic examination.
We will summarize the availability of key data needed for the primary analysis. All treatment assignments will be available and due to randomization of treatment assignment no covariates will be included in the primary intent-to-treat statistical analysis. The primary outcome variable is the change in pain scale from before first treatment (baseline) to 3 weeks after the last treatment, and the goal is to compare the mean change in pain between treatment groups. Should there be missing values in the primary outcome measurements of pain, we will modify the initial statistical analysis plan of a two-sample t-test for differences in the pain change score to an analogous linear mixed model, which provides estimates that are valid under a missing at random assumption.
BiasSeveral measures to reduce bias have been implemented, including that all questionnaires are self-reported and have the option of being provided in English or Spanish, with the option for the participant given the option of “I prefer not to answer” for any questions, due to the sensitive and intimate nature of some questionnaires. Standardized screening processes are utilized, which can be provided either in English or Spanish, depending on the potential participants’ preference to improve communication, understanding, and inclusivity.
While we are recruiting from various settings, one bias noted is that the majority of our participants captured from advertisements/flyers (ICD-10 code based) are those with healthcare insurance vs. those without health care insurance, which can be linked to differences in socioeconomic factors. Additionally, our study is recruiting females only, assuming they not only identify as female but have female anatomy; however, we realize this is a limit in our inclusion criteria description, as we are basing our recruitment on female anatomy and not gender.
留言 (0)