
記住我



See section Study Design.
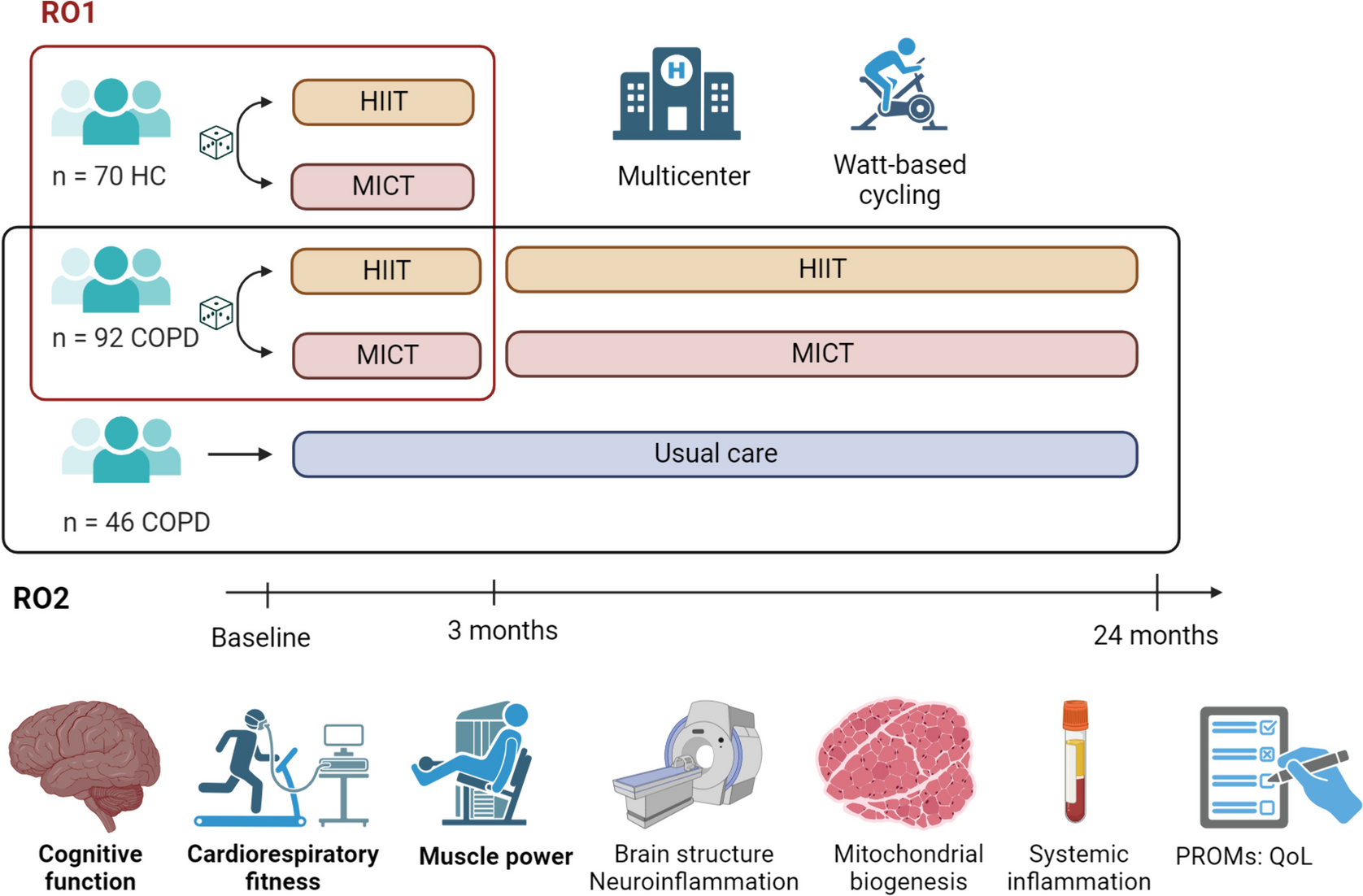
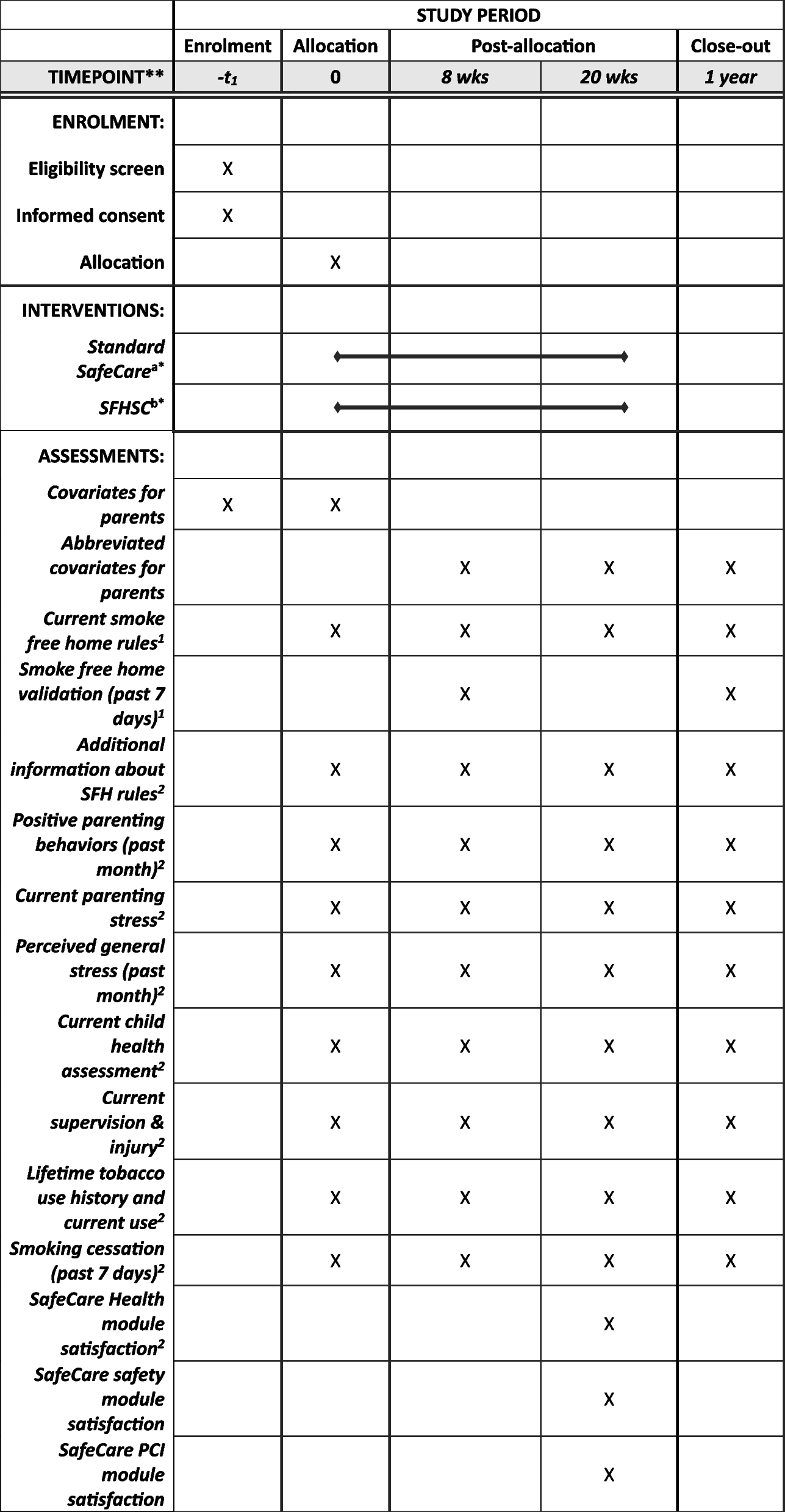
A description of the study flow is given in Fig. 1 whereas the study calendar can be found in Table 2.
Fig. 1 Table 2 Study timeline. V0 to V14 represent the succesive patient visits for the purposes of this study. V0 is the inclusion visit (see heading « Visit Schedule»), V1–V6 represent the first phase; V7 is the evaluation after the first phase and cross-over, V8–V13 are the second phase visits and finally V14 is the final visit
Table 2 Study timeline. V0 to V14 represent the succesive patient visits for the purposes of this study. V0 is the inclusion visit (see heading « Visit Schedule»), V1–V6 represent the first phase; V7 is the evaluation after the first phase and cross-over, V8–V13 are the second phase visits and finally V14 is the final visitThe total duration of the study is 45 months between the inclusion of the first patient and the end of follow-up of the last patient. This time includes the following:
Preparation of the study including ethics, regulatory, and legal consideration as well as opening of the 8 centers: 6 months
The recruitment of patients: 24 months (after ethics, regulatory, and legal authorizations)
One month between oral consent + psychological test and the beginning of the treatment
The 12 months of treatment with ziconotide or placebo + 1 month of wash-out
The end of the study is the date of the last follow-up visit of the last person participating to the study, or when the last patient’s last visit window is closed, whichever is the earliest.
SettingThe recruitment, treatment, and follow-up of patients will be carried out by investigators in the neurosurgical, or the algologist or the physical medicine and rehabilitation units participating to the study.
Visit scheduleIntrathecal pain therapy is routine treatment for patients with severe refractory pain and according to HAS recommendations ITZ may be proposed to patients with refractory neuropathic pain after a test period. Patients meeting eligibility criteria (candidates having tested positive to ITZ) will be proposed to enter this study by the participant pain clinics. The inclusion visit (V0) is therefore scheduled after the implantation of the IT pump. This is followed by visits 1–6 for the first phase of treatment with its initiation at V1 according to randomization and subsequent adjustments made at each FU visit from V2 to V6. At V7, the first phase of treatment ends with a detailed evaluation and the cycle is taken up again after crossover (V8 to V13). V14 is the final study visit. Routinely patients with SCI pain are tested for the efficacy of ITZ and the test phase is not the object of this study. However, some recommendations and suggestions are made:
Patients will be tested according to the test protocol. First, an evaluation of cerebrospinal fluid (CSF) circulation will be conducted with MRI. If a block or a suspicion of block in the CFS circulation is identified, patients will be tested with a continuous infusion test. The other patients will first be tested with an LP test. Patients considered responders will be the target population of this study.
A proposed protocol used during the pilot study is as follows:
LP testThree LPs are performed with a 72-h interval between. Boluses of ziconotide are administered with progressively increasing dosages. Doses are respectively 0.5, 1.0, and 1.5 µg diluted in 2 mL of saline. Patients are monitored for vital, neurological signs just before LP and every hour for the first 24 h after LP and then every 4 h for the subsequent 24 h. Biological values including creatin phosphokinase and creatinine are measured 24 h after the first LP and at the end of LP test period. The ward nurse assessed thereafter VAS and adverse effects (AEs – 6.1.3) during each visit (just before the LP and 1, 4, 8, 12, 24, and 48 h after the LP). After 24 h, patients are asked to grade their degree of pain reduction from 0 to 100% and their degree of satisfaction with the therapy.
Patients are considered responders if a reduction of VAS greater than or equal to 40% or if they declared a degree of satisfaction of more or equal to 40%. Responders to the LP test are implanted with a continuous infusion pump. Patients having severe adverse effects (AEs) during the test period are not implanted with a permanent pump as well as patients not desiring the therapy.
Continuous infusion testAn intrathecal catheter is connected to a subcutaneous small reservoir (see below for technique). An external pump (Cane Crono 5 Infusion Pump, Applied Medical Technology, and Italy) is connected to the subcutaneous site via a HUBER needle. Then a continuous infusion is performed at dosage from 2 to 10 µg maximum per day with an increment of 1 µg every 3 days. Pain is evaluated every 4 h for the VAS and every day for the degree of satisfaction. As for LP test patients are considered responders if a reduction of VAS greater than or equal to 40% or if they declared a degree of satisfaction of more or equal to 40%.
Some patients might already be implanted with a Syncromed II pump with Baclofen treatment. These patients will be proposed to participate to the study and go through the testing phase using the pump (continuous infusion test).
Finally, according to local practices, the neurosurgeon can decide to implant directly the Syncromed II pump for the pre-test phase (according to the procedure below). This allows a more accurate infusion dose, in case of failure to reduce of VAS greater than or equal to 40% or if they declared a degree of satisfaction of more or equal to 40%, the pump can be used for Baclofen treatment and in case of success, the pump is already implanted for the study. This procedure is not study related but is becoming a standard of care in several centers, starting with Lyon.
Pump implantation technique is at the choice of the neurosurgeon and respect the current practice. Detailed recommendations for the implant technique are found in the supplementary material.
For patients receiving IT analgesics, a pump with 40-mL reservoir is preferable. But under special conditions, a smaller 20-mL pump may be acceptable. If a smaller pump is used, the steering committee should be advised to adapt the treatment protocol.
Details pertaining to the pump implantation are as follows.
Before intrathecal infusion testing, the subarachnoid space may be checked for absence of CSF blockage by T2 MRI sequences of the entire spinal or by myelography. For these patients, the intrathecal catheter should be implanted above the lesioned level. In all other patients, the catheter may be implanted in the lumbar region with the tip facing the conus medularis whatever the lesional level.
The pump must be placed at the right depth. If the skin coating is not thick enough, the pump implantation must be under fascia.
Lumbar catheters are generally implanted using a percutaneous technique with a TUOHY needle. The position of the tip was verified by intraoperative radiology. In case of blockage, supralesional catheter is placed surgically trough an interlaminar approach. A midline incision of the dura is performed and the catheter was passed in cranial direction with the tip placed two or three vertebral levels above the lesioned level. A circular suture is made to fix the catheter to the dura and ensure watertight closure. An injection site is placed subcutaneously in the abdominal region (usually on the left flank) and connected to the catheter after checking the CSF flow.
Permanent, subcutaneous continuous infusion pumps are implanted in cases were LP tests or continuous infusion tests are positive. A Syncromed II pump by Medtronic Inc., WI, USA, is to be implanted. Both the 20-mL (ref 8637) and the 40-mL (ref 8637) pumps may be used according to the dosage used during testing and patient morphology (see above).
The study population is the population of patients having tested positive to an LP test or a continuous infusion test. Patients that are non-responders are not to be implanted according to HAS recommendations and are not to be included in SPIDOL study. They are readdressed to their respective pain centers for alternative pain therapies.
Patients selected for the study will meet the neurosurgeon to propose the intrathecal treatment and inclusion in the study. Non-inclusion in the study does not preclude ITZ. He explains its purpose, both strategies studied, the randomization process, the objectives, the risks and benefits, and the follow-up visits following treatment strategy. He gives them a written information letter containing all these elements and a participation consent form. After a time of reflexion of 15 days corresponding to the wash-out phase of the pre-test needed before the psychological test, the patient will be pre-included in the study if he agrees to consent (written informed consent).
Pre-inclusion visitFor patients corresponding to the inclusion criteria and having given written informed consent, exclusion criteria will be tested with the Mini International Neuropsychiatric Interview questionnaire (MINI) to evaluate main psychiatric disorders. Tobacco consumption, dysthymia, recreational or therapeutic cannabis consumption, video games or gambling are not included in the MINI evaluation or as criteria of exclusion as these addictions will not interact with the PRIALT® prescription. However, the neuropsychologist in charge of the patient evaluation can decide, on any of these criteria, to exclude the patient if an addiction or a behavior that can interact with the PRIALT® prescription is identified.
The inclusion visit will be performed for patients without any psychiatric disorders tested by MINI. At this visit a first full assessment will be performed:
◦ Exact localization of the spinal cord injury
◦ Full neurologic workup
◦ Sensory deficit chart
◦ Pain distribution chart
◦ Overall pain at the moment of assessment (VAS), pain estimation over the past week (VAS – Baseline) using an electronic pain diary
◦ Pain description in the following terms: at level/below level pain, continuous/paroxystic pain with quantification of each (VAS)
◦ Quantification of daily dosages of analgesic drug intake with differentiation of class of analgesics
◦ Quality of life scale (SF12)
◦ Mc Nair scale for memory complains
◦ HADS scale for depression and anxiety
◦ BRIEF-A questionnaire for impact on behavioral manifestations of executive functions
◦ PCS to assess the patient’s physical and emotional distress associated with their pain condition
All this information will be re-evaluated at the end of each study phase (V7 and V14) and at the wash-out phase.
After giving his consent passing through the MINI test, the inclusion visit, and the pump implantation, randomization is performed. Randomization will be centralized and balanced between each participating center.
The group assignment of an included patient will be based on the chronological order of entry into the study according to a predetermined randomization list. The randomization list will be only detained by the biostatistics unit, the local pharmacy, and the CAP.
The randomization procedure will be carried out by the investigator using a secure and dedicated web server. The coordination center of the study will be alerted by email of the patient inclusion and of the following information: the patient initials, his date of birth (month and year), and the inclusion date.
A number is allocated to the patient, corresponding to a couple of prescription (placebo and ITZ) in a specific order. This prescription is sent to the hospital pharmacy of each center which will then deliver the anonymized treatment.
1)Experimental treatment: Ziconotide
Ziconotide is currently available with a concentration of 100 μg/mL. Marketed vials have volume of 1 or 5 mL solution for infusion. If dilution is required, ziconotide must be diluted aseptically with preservative-free sodium chloride 9 mg/ml (0.9%) solution.
The experimental treatment in SPIDOL consists of ziconotide solution that will be prepared by each local pharmacy team, in 5- or 10-mL vials with constant concentration of 10 μg/mL. So, the daily dosage will be set only by control of the injected volume. This strategy using a solution with constant concentration facilitates the blinding of the solution injected. Moreover, the 10 μg/mL dosage allows to optimize the consumption of the solution. Pumps will be refilled every 30 days. Initial dosage will be established by the treating team according to the response to the ziconotide pre-test. An increase in dose may be performed at any of the refill visits. Interim visit between refill visits for only dose adjustment is allowed once only to avoid too fast increase of daily dosage. The magnitude of increase is the decision of the treating physician (as long as it remains with the recommended limits by the HAS) but most likely augmentations will be at maximum 1 μg per month and maximum achieved doses allowed in SPIDOL study is 20 μg/day.
To use a minimum amount of drug, pumps will be filled according to the volume injected per thirty-day period with 10, 20, 30, or 40 mL of product. In this way, the maximum possible dose is 13.3 μg per day. In order not to cap the maximum dose, patients requiring higher doses should be seen more frequently than 1 month. Refill volumes should be as follows:
For dosages above 13.3 µg per day, the steering committee should be consulted.
2)Placebo:
The placebo treatment consists in standard saline solution (preservative-free sodium chloride 9 mg/ml (0.9%) solution) which will be presented exactly in vials as presented for the treatment (volume, color, shape, and size of the vial). Treating physicians will not be aware of the actual contents of the pump and will increase the volume of injected placebo as a treatment solution (as ziconotide 10 µg/mL).
1)First phase (6 months) (V1 to V6)
Two weeks after pump implantation the treatment is started at a dose determined during the test. Initial dose of around 3 µg per day is advisable. It is advisable to start the infusion with a short hospital stay. This can be also performed through several visits during the first week of treatment. Further increases in dose should be performed either at pump refill visits or at interim instance between refills. The duration of the first phase is 6 months of treatment.
Patients having been tested through continuous testing and responding to doses higher than 3 µg per day, should be initiated at the efficient dose determined by the test. Sufficient hospital length of stay or outpatient clinic visits should be provided to initiate safely such a dose. Same speed of increase of the dose as during the test phase should be used but the recommendation is to use a step of 0.5 µg per day after an initial start at 3 µg per day until the efficient test phase dose is reached.
Each month after the initiation of the first phase of the treatment, a visit is organized to refill the pump with the prescription corresponding to the random first phase and a pain evaluation is performed.
At each of these monthly pump refill visits, a VAS pain evaluation is conducted, MINI are filled in by the patient and AE are collected.
At the end of the last dose of treatment of the first phase the pain level is evaluated using a numeric scale and all the other assessments are performed by blinded physicians.
Initial dosage increases and visits for refill and follow-up are in concordance with usual scheduled visits for patients receiving intrathecal ziconotide or other pain IT treatment as routine treatment.
Schedule for initial doses and follow-up visits during treatment phase:
Treatment initiation:
Titration to calculated dose during the test phase:
◦ Short hospital stay preferable
◦ Incremental increase to the desired dose (i.e., effective test dose) by 0.5 µg per day
◦ For doses above 1 mL per day steering committee should be consulted
◦ Dose titration will be halted at the effective dose determined during the test period (regardless of result—since some patients receive placebo)
◦ After the end of dose titration, the pump may require a refill to ensure sufficient drug for the duration up to the next refill visit—this is allowed at each visit
Refill visits each month after the initiation of the treatment. The choice of reducing the refill interval to 1 month is to avoid any product decay in the interim period and is formal, 6 refill visits must be scheduled at 1-month intervals after the initiation period of each phase.
◦ Each day the patient assesses his/her mean pain on an electronic dairy
◦ At each refill visit, patients will be tested by the blinded physician for pain scores, screened for adverse effects.
◦ Pump refills are performed in the same fashion as refills for other patients requiring IT treatment as per center habits.
◦ The refill agent will be prepared in pharmacy at the previsioned volume for that instance with allowance for dosage increase at that visit or at a potential interim visit.
An interim visit to adjust dosage on patient demand may be scheduled between the visits. A single visit is allowed—since side effects are in relation with the speed at which the dose is increased. The amount of increase is left to the choice of the treating physician as long as it does not surpass the HAS recommendations. The advised dose increase is 0.5 µg per day in the case of active treatment.
The last refill visit is the end of study phase visit. At this visit, the pumps will be filled with serum saline for the wash-out period and all primary and secondary outcome tests will be performed.
A wash-out period of 1 month is organized to rule out the potential carry over effect; during this period, the pump is filled with preservative-free sodium chloride 9 mg/ml (0.9%) solution (allowing flushing the dead space of the catheter). All primary and secondary outcome measures and tests will be performed at the end of the wash-out period before the initiation of the second phase of treatment.
The second phase also last for 6 months. The second intrathecal infusion is initiated with refill of the pump with the random second preparation (placebo or ITZ) on the exact same protocol in terms of refill visit and questionnaires as the first phase but with the other product compared to the first phase. An initiation period of the second phase will be purported just as for the first phase with exactly the same schedule for visits. The last pump refill visit will occasion the end of study visit at which point all outcome measures will be again performed (Table 3).
Table 3 Pump refill volumesAll the assessments are performed by blinded physicians; in fact, all physicians are blinded to the presence or not of active treatment. Unblinding occurs after the completion of the end of study visit initiating the post follow-up treatment.
During both phase of treatment each patient will be asked, on a daily basis, to evaluate his pain using a VAS scale. This data will be collected directly on the eCRF thanks to a patient-specific access with a username and password. In order to respect the confidentiality (MR001 CNIL), the access code will be transferred by the data manager of the study to the patient via his physician or Clinical Research Associate. The Clinsight software will be used for this study.
This will allow the pain evaluation to be as accurate as possible and this will participate to reduce the attrition risk of the study.
All along the study, the patient will be able to take all available analgesic treatments except interventional therapies (infiltrations, botulinum toxin, Cutenza, neurostimulation in all its forms).
During the last visit, the patient will be informed in which period he was under active treatment and may choose whether to continue/reinitiate active treatment with ziconotide. The patient will take the appropriate decision after discussion and agreement with his physician. Those deciding to continue to receive the drug will be followed as similar patients receiving IT treatment (for ziconotide once a month refill visits) but outside the study frame. The optimal ziconotide dose observed during the study will be proposed for further use. This will be considered the minimal dose at which the patient had the greatest decrease in pain score without severe adverse effects or intolerable side effects (as defined by the patient). Patients not choosing active treatment will be readdressed to the pain center for testing of other treatment strategy.
Blinding methods and unblinding procedureBlinding procedure will be systematic thanks to the indistinguishable nature of the active product and placebo and their packaging. It will be organized by each local pharmacy of the participating centers.
Only the statistician in charge of the production of the randomization list, the Centre Anti-Poison of Lyon, and the authorized pharmaceutical team responsible for packaging, labeling, and distribution of TU to the sites will have access to a decoded list.
Unblinding procedure will be possible 24 h/24 h, 7/7 days, simply by phone call to the Centre Anti Poison de Lyon (CAP—04 72 11 69 11). The CAP physician will be able to proceed to the unblinding if required upon request of investigator. Unblinding will be reserved for clinical conditions where study treatment knowledge is likely to influence the management of the adverse event.
A participation card will be provided to subjects enrolled in the study, including the telephone number of the CAP, as well as the information necessary for the unblinding request.
Study calendarSee Table 2 for the study calendar.
Temporary or permanent terminationAny termination of study participation such as ceasing to follow-up for the study or early withdrawal should be clearly documented in the eCRF. The termination of normal follow-up in the clinical study is when the patient completes the visits after the second phase of treatment. The participation of a patient will be permanently stopped and the patient will be considered prematurely withdrawn from the study in the following cases:
◦ Withdrawal of consent at any time during the study, without any explanation and without penalty or prejudice to the patient’s healthcare, as required by Authority Regulation.
◦ Presence of an exclusion criterion (prior to randomization);
◦ Violation of a protocol, defined as any event violating the patient’s right, safety or well-being, or affecting the integrity of the research (including the non-compliance with eligibility criteria);
◦ Upon decision of the investigator, in case the patient is lost to follow-up despite several attempts to contact him. The death record of the patient will be searched.
◦ In the investigator’s opinion, if further participation in the study would be detrimental to the subject’s well-being.
◦ CPK increased > 5 ULN, or associated with clinical signs of myopathy or rhabdomyolysis
◦ Infection of pump site or scar
◦ Pump malfunction
In case of withdrawal of consent, no additional data will be included in the study database. Data already included will be kept and used for data analysis. If the patient decides not to withdraw his consent, data from his normal follow-up will be collected in the database.
These subjects will not be replaced, as study drop-outs have been included in the calculation of the sample size.
The research can be temporarily or permanently stopped for the following reasons:
◦ Upon decision of the coordinating investigator, the sponsor or the competent authority;
◦ In case of the knowledge of data jeopardizing the achievement of the study due to the patient safety;
◦ In case of publication of new scientific data questioning the research;
◦ In case of serious adverse events which DSMB recommends the termination of the research, as it considered it (a) serious and unexpected, (b) involving the safety of the patients, and (c) suspected to be related to the research.
◦ On request of Health authorities
Premature discontinuation of study treatment is not considered as a study drop-out. In accordance with the principle of intention to treat, all randomized subjects who received a treatment unit will be considered in their allocated treatment group in the analysis.
Identification of the data to be collected directly into the case report formThe documentation of the inclusion and non-inclusion criteria will be made directly by the examiner using a specific form to be considered as a source document. For each patient, this form will be filed in the CRF binder.
Unblinding procedureIn case of serious adverse events (SAE), unblinding procedure will be organized by the pharmacovigilance service. If the distribution of SAE is unbalanced, the DSMB will be alerted and will then determine the relationship between the type of treatment (ziconotide or placebo) and the occurrence of the event.
Unblinding procedure required by the physicians will be restricted to the patient management modifications.
Whatever the causal relation selected, the patient, regardless of the randomization group, will not be excluded from the study and will be followed up according to the protocol. This deviation from the protocol will be collected and documented.
Experimental treatment given to study participantsExperimental groupThe medical treatment concerned by this research project is the ziconotide PRIALT®.
Control groupThe control group will be a placebo (preservative-free sodium chloride 9 mg/ml (0.9%) solution) with the exact same form, size, and taste as the experimental treatment.
Other treatment used for research purposesNo other treatment is used for the purposes of the study (no procedure other than those used in the treatment current practice of these patients).
Permitted and prohibited treatmentsProhibited treatment during the study will be instrumentation not specified in the experimental/control group and clonidine, bupivacaine, and propofol. As propofol is a frequently used anesthetic, its prohibition will be indicated in the information notice. The anesthetist should then use another anesthetic.
Considering the contraindications to the use of ziconotide PRIALT®, women of childbearing age should have effective contraception (oral contraception or intrauterine device or contraceptive implant). Contraception should be maintained throughout study participation.
Statistical analysisDescriptive analysisA descriptive analysis will be performed on all recorded quantitative (average, median, max min, quartile) and qualitative (frequency and percentage) parameters with their associated confidence level.
The hypothesis of normality of distribution for the quantitative variables will be verified graphically with a histogram and statistically with the Kolmogorov–Smirnov test. Log transformation or outliers’ exclusion might be used if necessary.
The intention to treat population (including subject randomized according to randomized treatment assignment, regardless of noncompliance, protocol deviations, withdrawal, and anything that happens after randomization) and the per protocol population (patients completing the study protocol without any major protocol violations) will be described in detail.
Treatment of missing, unused, or invalid dataAll data regarding the primary endpoint will be used for statistical analysis. The missing or invalid data regarding the other endpoints will be reviewed by the principal investigator and the person in charge of the data analysis. They will decide if the data can be taken into account in the analysis or not. Multiple random imputations might be used to replace or complete missing data.
Protocol deviationA description of protocol deviation (lost to follow-up, non-compliance with trial drug, problem with the pump…) which could interfere with the interpretation of the results, and be considered as potential bias will be realized. The reasons for protocol deviation will be analyzed.
Intermediate analysisNo intermediate analysis will be performed upon the primary endpoint so as to keep sufficient statistical power.
Carry over effect evaluationIn order to check the assumption of a negligible carryover effect, a pre-test will be conducted according to Wellec et al. (2012). In case the carry over effect exist between the first and second phase, then the analysis will be restricted to the first phase of the study and conducted as if the study was 2 parallel groups.
Main criteria analysisThe main analysis will concern the intra-individual comparison of the average pain measured under placebo and under ziconotide using a numeric scale.
First, after confirmation for a negligible carry over effect a paired T-test or a Wilcoxon signed rank test, according to the normality of the distribution, will be conducted to compare the intra patient treatment effect between the 2 sequence of treatment allocation.
Second, being repeated measures (daily basis pain evaluation with VAS), the treatment effect on pain will be estimated by a mixed-effect linear model (Proc Mixed, SAS) to account for the repeated measurement that yield period, sequence, and carryover effect. It will also allow us to model the various sources of intra patient and inter patient variability. A first step will be to evaluate the last 2 weeks of treatment: indeed, there is a long period of dose adaptation and stabilization. Secondly, an evaluation on the whole treatment period will be conducted.
Secondary criteria analysesThe following secondary analysis will be conducted:
Proportion will be compared in between the 2 groups using a chi-square test or a Fisher exact test if the conditions of application of chi-square test are not met.
On the exact same process as describe above for the primary endpoint, repeated quantitative variables such as SF12, McNair, PCS, HADS, BRIEF-A, satisfaction, duration, and intensity of provoked pain, continuous – paroxysmal pain will be compared between treatments using mixed-effect linear model [35, 37,38,39].
Modification of statistical planThe information presented above constitutes the basis for the statistical analysis plan for this study. This plan may be revised over the duration of the study in order to accommodate any amendments to the clinical trial protocol or adapt to any unforeseen difficulties in carrying out the study, which could impact the planned analysis.
The plan will be edited before the review of the data if needed; any revisions will be made before the database freezing. The analyses provided may be completed during this review. Thereafter, any changes to the analysis plan will result in a new version, including justification for the changes. All will be archived in the study file. The final statistical analysis plan will be made available on request from the study organizers and as supplementary material for the final publication if not published as a standalone text prior to this.
Statistical softwareThe SAS Institute Inc. software version 9.4 will be used to perform all analyses according to the programs and the applicable procedures within the clinical research unit.
The statistical tests are bilateral, and the level of significance was set to 5% (p < 0.05).
Statistical analysis unitData management and statistical analysis will be conducted by the Unité de recherche et d’épidémiologie Clinique du pôle de santé publique des Hospices Civils de Lyon.
Data quality insuranceSource document requirementsAccording to the guidelines for Good Clinical Practices, the study monitor has to check the case report form entries against the source documents. The Informed Consent Form will include a statement by which the patients allow the sponsor’s duly authorized personnel (trial monitoring team) to have direct access to original medical records which supports data on the electronic Case Report Form (e.g., patient’s medical file, appointment books, original laboratory records). These personnel, bound by professional secrecy, will not disclose any personal identity or personal medical information (according to confidentiality rules).
Case report formThe case report form will only include the data necessary for an analysis for a scientific publication. Other patient data necessary for their follow-up outside of this study will be collated in their medical file.
A specific online module of the electronic CRF available on a smartphone is proposed to the patients so that they can provide daily information about their pain at home.
Electronic CRFAll information required by the protocol will be recorded in the case report form using the electronic CRF Clinsight from ENNOV Clinical (Paris, France ennov.com). Data must be collected as it is obtained and explicitly recorded in these case report forms. All missing data must be encoded. This electronic case report form will be put in place in each center through an internet portal for recording the data. A help document for using this tool will be provided to the investigators.
The completion of the case report form by the investigator through the internet allows the study coordination center to rapidly see the data at a distance. The investigator is responsible for the accuracy, quality, and pertinence of all the data entered. Furthermore, during entry, these data are immediately verified thanks to coherence checks. As such, the investigator must validate any value changes in the CRF. These changes are part of an audit trail. A reason may be optionally integrated as a comment. A print-out will be requested at the end of the study, authenticated (dated and signed) by the investigator. A copy of the authenticated document destined for the sponsor must be archived by the investigator.
Data managementThe Department of Clinical research and epidemiology will be responsible for data processing in accordance with their data management procedures. Data will be entered electronically via a web browser. A data backup Twice-daily for the Ennov Clinical server, hosted at OVH, as well as a Twice-daily data backup for Hospices Civils de Lyon servers is done. On the eCRF, an audit trail records connections/disconnections (as well as connection attempts) of all users + changes to data (who, when, what, why).
The sponsor will also organize a data monitoring with the clinical research assistant who will be trained on the clinical aspects by the PI, and on the use of the electronic CRF by the data unit.
Archiving clinical trial filesThe investigator shall maintain the essential clinical study documents (including source documents, clinical device accountability records, signed subject information consent forms, AE reports, and other regulatory documents) as required by the applicable regulatory requirements. The investigator should take adequate measures to prevent accidental or premature destruction of these documents. In the event of accidental destruction, the investigator must notify sponsor immediately.
The following documents will be archived under the name of the study and under the responsibility of the coordinating investigator or associated investigators in each site for 25 years.
Signed informed consent documents for all subjects.
Subject identification code list, screening log (if applicable), and enrollment log;
Record of all communication between the investigator and CPP;
Composition of the CPP or other applicable statement.
Record of all communications between the investigator and the sponsor
List of sub-investigators and other appropriately qualified persons to whom the Principal Investigator has delegated significant trial-related duties, together with their roles in the study and their signatures.
Copies of CRFs pages and of documentation of corrections for all subjects.
Device-accountability records.
All other source documents (i.e., subject records, hospital records, laboratory records);
All other documents as listed in Section 8 of the consolidated guideline on GCP (Essential Documents for the Conduct of a Clinical Trial). Essential clinical study documents shall be retained for at least 15 years following the date of the end of the study.
These documents shall be retained for a longer period, however, if required by additional applicable regulatory requirements or by an agreement with the sponsor. The investigator must, therefore, obtain approval in writing from the sponsor prior to the destruction of any records.
The investigator shall notify the sponsor to any change in the location or status of any essential, clinical-study documents. The sponsor shall be responsible for informing the investigator when these documents no longer need to be retained.
The sponsor is also responsible for organizing the storage of the statistical analyses and the final study report for the required duration of archiving.
No moving or destruction can be carried out without the agreement of the sponsor. At the end of the 25 years, the sponsor will be consulted for the destruction. All data, documents, and reports may be the subject of an audit or inspection.
留言 (0)