
記住我



Milled splints are used as comparators as subtractive manufacturing represents the standard CAM fabrication technique for splints. Another option would have been to use splints fabricated conventionally by alginate/silicone impression, cast fabrication, wax-up, and molding from powder-liquid mixtures of PMMA as comparators. It was decided to use milled splints as a comparator since the digital procedure will replace conventional processes due to better time-efficiency and patient comfort [18,19,20]. In addition, the comparison of two splints fabricated using different CAM processes enables the comparison of splints based on the same digital design and therefore allows differences in splint design/shape to be ruled out as the cause of possible differences in clinical outcome. In contrast, conventionally fabricated and digitally fabricated splints would differ in splint design/shape rendering direct comparison of the effects of the fabrication mode on clinical outcome infeasible.
Intervention descriptionParticipants are examined and diagnosed following standard protocols of the DGFDT and the DC/TMD (T0). The BSI, symptom questionnaire of the DC/TMD, the DC/TMD examination form, and the GCPS are used for this purpose. Afterwards, digital impressions of the participants’ upper and lower jaw are taken with an intraoral scanner (Trios 4, 3Shape, Kopenhagen, Denmark), and therapeutic relation, often entitled “centric” relation, is registered as described by Türp [26] with wax sheets at the intended height of the splint (in most cases approx. 2 mm interocclusal space in molar regions). Lateral bite scans are then performed with the intraoral scanner and the adapted wax sheet in situ. Based on this data set, the Michigan-type stabilization splints are designed with the software SplintStudio (3Shape). Then, based on this design, one occlusal splint is 3D printed from Primeprint splint (Dentsply Sirona, Bensheim, Germany; the material is marketed by Dentsply Sirona under the name Primeprint Splint, but is manufactured by DETAX, Ettlingen, Germany as Freeprint Splint 2.0, i.e., Primeprint splint and Freeprint Splint 2.0 are the same materials and both have the UDI DI EDET0465W). The 3D printing process is carried out using a Primeprint printer (Dentsply Sirona). Subtractively manufactured splints are milled from Ceramill A-Splint blanks (Amann Girrbach, Pforzheim, Germany) with an inLab MC X5 (Dentsply Sirona, Bensheim, Germany). The materials are processed as specified by the manufacturers. Participants are instructed to wear the splints at night.
Starting with the incorporation of the first splint, the trial is carried out according to the following protocol:
Incorporation of the 1st splint (T1): scan of the splint as delivered from the laboratory with the intraoral scanner and—if necessary—also after adjustment, completion of the Oral Health Impact Profile (OHIP-G14, German version from 2005) by all participants of both cohorts, completion of the GCPS by participants of the TMD cohort, examination of the TMD patients according to the DC/TMD standard
Control 2 weeks after incorporation of 1st splint (T2): if necessary, adjustment and scan of the splint, completion of the OHIP-G14 and GCPS and examination of TMD patients as described above, rating of wear comfort and retention of the stabilization splints on a VAS scale of 1–10 by participants of both cohorts
Control 3 months (11–13 weeks) after incorporation of the 1st splint (T3): scan of the splint and opposing dentition with the intraoral scanner, completion of the OHIP-G14 and GCPS, examination of TMD patients and rating of wear comfort and retention as described for T2, scan of the 2nd splint as delivered from the laboratory and—if necessary—also after adjustment, incorporation of the 2nd splint.
Control 2 weeks after incorporation of 2nd splint (T4): see T2
Control 3 months (11–13 weeks) after incorporation of the 2nd splint (T5): see T3. Additionally, the participant is asked to name his or her favorite splint (1st or 2nd)
A wash-out period between the two splints was not included, as this may lead to aggravation of symptoms and therefore bears the risk that patients may switch to alternative treatment options such as commercially available splints to alleviate symptoms during the wash-out period, rendering trial conditions less controlled.
Criteria for discontinuing or modifying allocated interventionsCriteria for discontinuing or modifying allocated interventions for a given trial participant are the occurrence of exclusion criteria for the respective cohort or the withdrawal of consent. Interventions may also be discontinued if a participant unexpectedly experiences a worsening of symptoms that the participant considers intolerable.
Strategies to improve adherence to interventionsFrom the strategies to improve adherence in clinical research proposed by Robiner [27] and Matsui [28], the following aspects are considered in this trial: the study personnel spends sufficient time during the first examination to assess the participants’ potential adherence. The protocol, the benefits for (future) patients, and the importance of adherence are explained in detail to participants to help them understand the objectives of the trial and the importance of their adherence. Participants are explicitly informed about when and how to wear the splint. The appointments are scheduled at convenient times and the splints are offered free of charge to compensate for the time required to participate in the trial. Adherence is monitored by self-report of the participants. In general, participants can be assumed as intrinsically motivated to wear the splints, since this will reduce teeth abrasion and/or pain.
Relevant concomitant care permitted or prohibited during the trialDuring participation in the trial, participants should not undergo concomitant treatments or interventions like botulinum toxin injections into masticatory muscles, pain therapy with prescription painkillers, muscle relaxants, or physiotherapy. If any of these measures are necessary for medical reasons, further participation in the trial will be discussed on an individual basis. Short-term use of over-the-counter painkillers is permitted as well as application of heat/cold, self-massage, and continuation of physiotherapy exercises already learnt prior to the trial.
Provisions for post-trial careOnly established treatment modalities with a very low risk for adverse events are applied and participants can remove the splints themselves when they feel uncomfortable with them. Harm induced by trial participation, therefore, appears improbable which is the reason why no compensations for harm suffered from trial participation are deemed necessary. Participants can keep both splints after trial participation and are invited to regular controls at our department.
Outcomes Primary outcomesAs the primary outcome, the OHRQoL of the participants is assessed using the OHIP-G14 questionnaire on the day of incorporation and after 2 weeks and 3 months after incorporation of each splint. Depending on the answer options ticked by the participants, this questionnaire results in scores between 0 and 56. For each participant and splint, the difference from the baseline (incorporation of splint) is evaluated at 2 weeks and 3 months post insertion.
Additionally, participant satisfaction with the stabilization splints is investigated by asking the questions “How comfortable was wearing the splint for you on a scale from 1 (very uncomfortable) to 10 (very comfortable)?” and “How stable did the splint rest in your mouth at a scale from 1 (very unstable) to 10 (very stable)?”. These questions are asked at the 2 weeks and 3 months controls and are analyzed for differences in relation to the manufacturing method.
Secondary outcomesSecondary outcomes are treatment efficacy and technical results of the stabilization splints. In the present trial, treatment efficacy is defined as the extent of abrasion of the opposing dentition in the bruxism cohort since the reduction of teeth abrasion is the therapeutic rationale for splint incorporation in bruxers. Teeth abrasion is measured as volume loss of the opposing dentition by superimposing intraoral scans obtained at digital impression taking and 3 months control appointments. Treatment efficacy in the TMD cohort is additionally investigated by quantification of pain reduction as the lead symptom of pain-related TMD. For this, the GCPS is evaluated on the day of incorporation and control appointments. The differences in the values obtained for chronic pain intensity (point score between 0 and 100), disability days (between 0 and 30), and interference score (point score 0–100) at control appointments and incorporation days are then calculated for every participant and splint type. Further, the number of painful points on examination according to the DC/TMD is compared at control appointments versus the incorporation date (range 0 to 48). Jaw mobility is investigated by measuring maximum jaw opening, laterotrusion, and protrusion in millimeters at incorporation and control appointments.
In addition, the technical results of 3D printed and milled stabilization splints are compared. Fit at incorporation is evaluated by calculating the amount of adjustment necessary in µm3 by overlaying scans before and after adjustment of the splint. Wear of the occlusal splints is assessed by calculating volume loss in µm3 by overlaying scans of the splints obtained at incorporation and the 3 months control. The integrity of the occlusal splints (no fracture of the occlusal splint) is assessed at control appointments, and the rate of fractures occurring is calculated in percent for each type of splint.
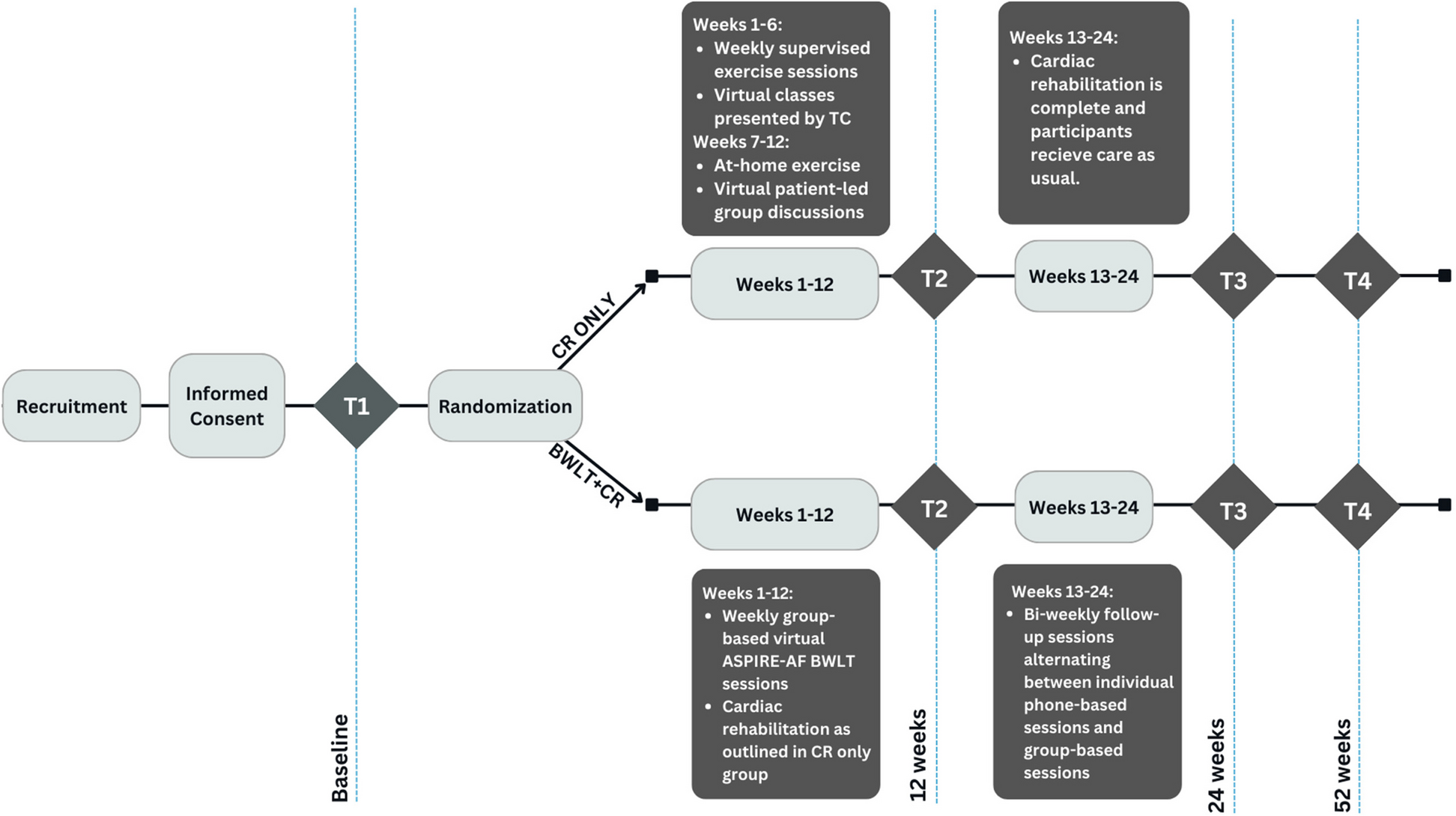
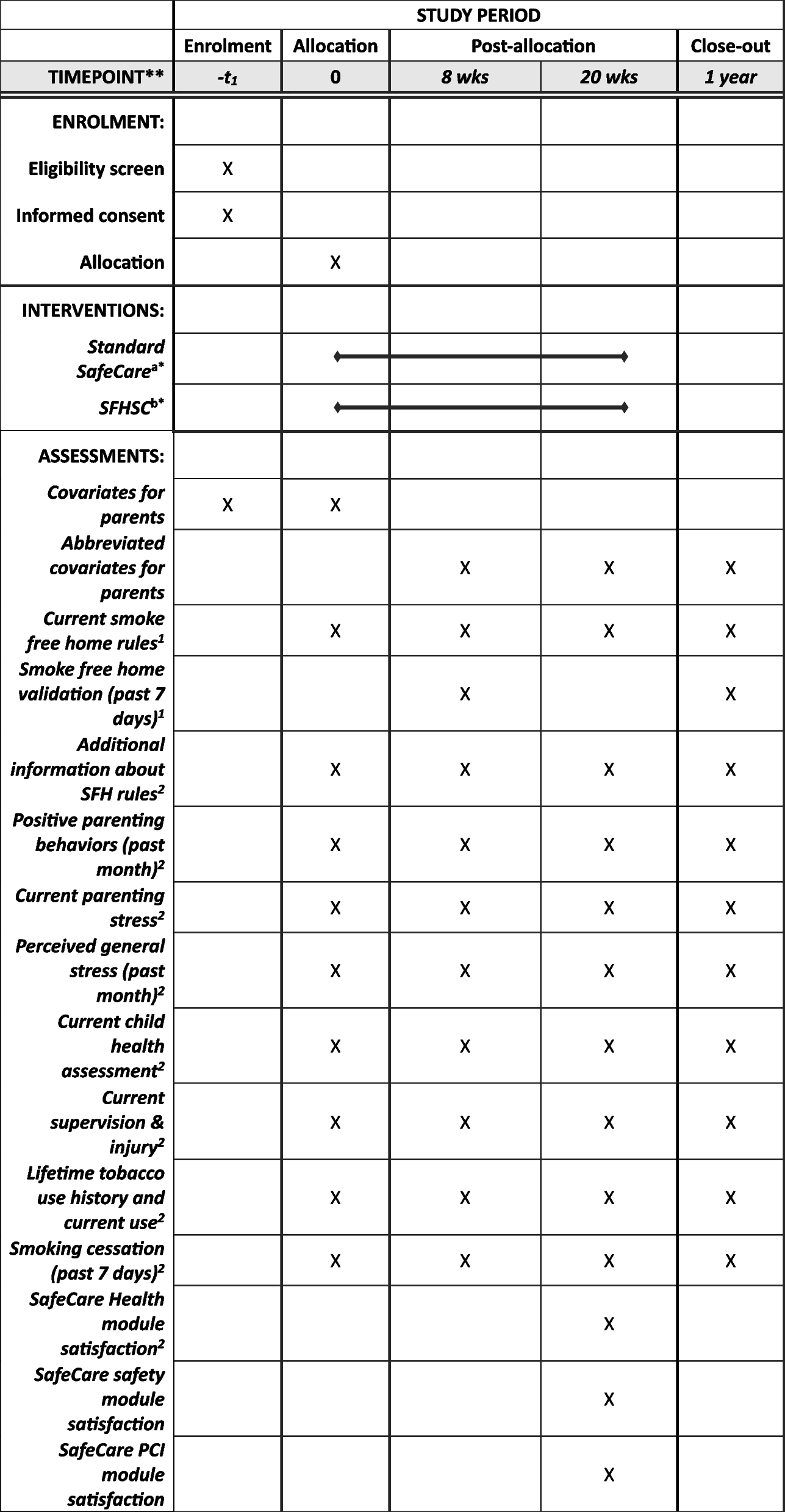
Participant timelineThe participant timeline is depicted in Fig. 1. Additionally, Fig. 2 illustrates the standard protocol items diagram according to the guidelines proposed by Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT).
Fig. 1
Participant timeline. Sequence of splints (3D printed or milled) is randomized at an allocation ratio of 1:1
Fig. 2
Standard protocol items diagram according to the SPIRIT guidelines
Sample sizeThe sample size was calculated based on results reported for the OHIP-G14. According to the results of Alajbeg et al. [29], an OHIP-G14 score of 25.29 (SD 12.38) can be assumed for TMD patients. If the non-inferiority margin is set at 5 and a standard deviation of the differences of 10 is assumed, 38 participants are required for the trial with a two-sided paired t-test and a power of 90% on a 5% significance level. As both cohorts can be analyzed together for the primary outcome parameter OHIP-G14, the target number of cases is set at 20 participants per cohort, resulting in 40 participants in total.
RecruitmentDentists of the Department of Prosthetic Dentistry, Medical Centre, University of Freiburg, Germany screen patients for trial eligibility. No further strategies for achieving adequate participant enrolment are conducted to reach the target sample size since both—bruxism and TMD—are frequent in the patient population of the above-mentioned clinic.
留言 (0)