
記住我
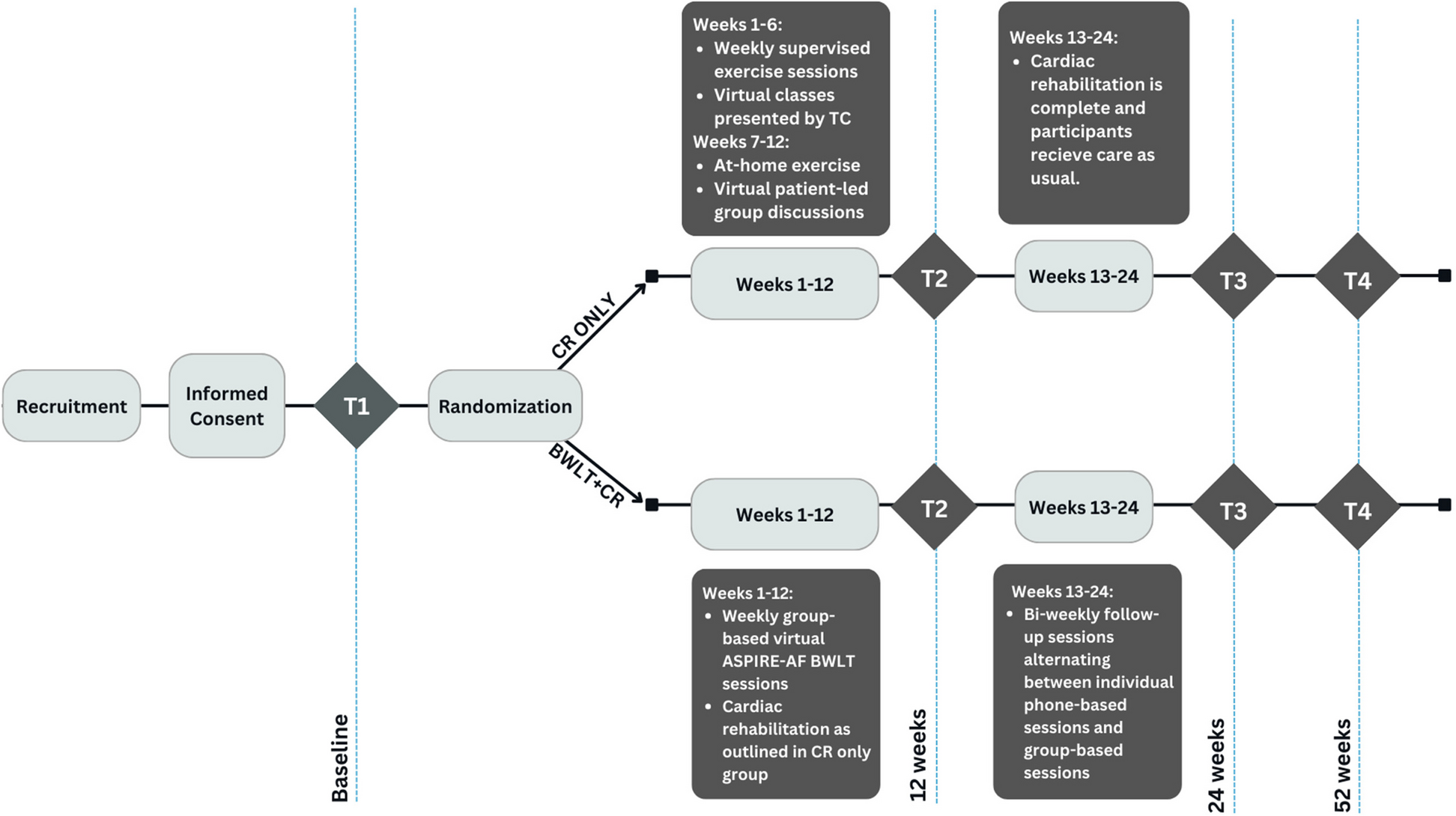

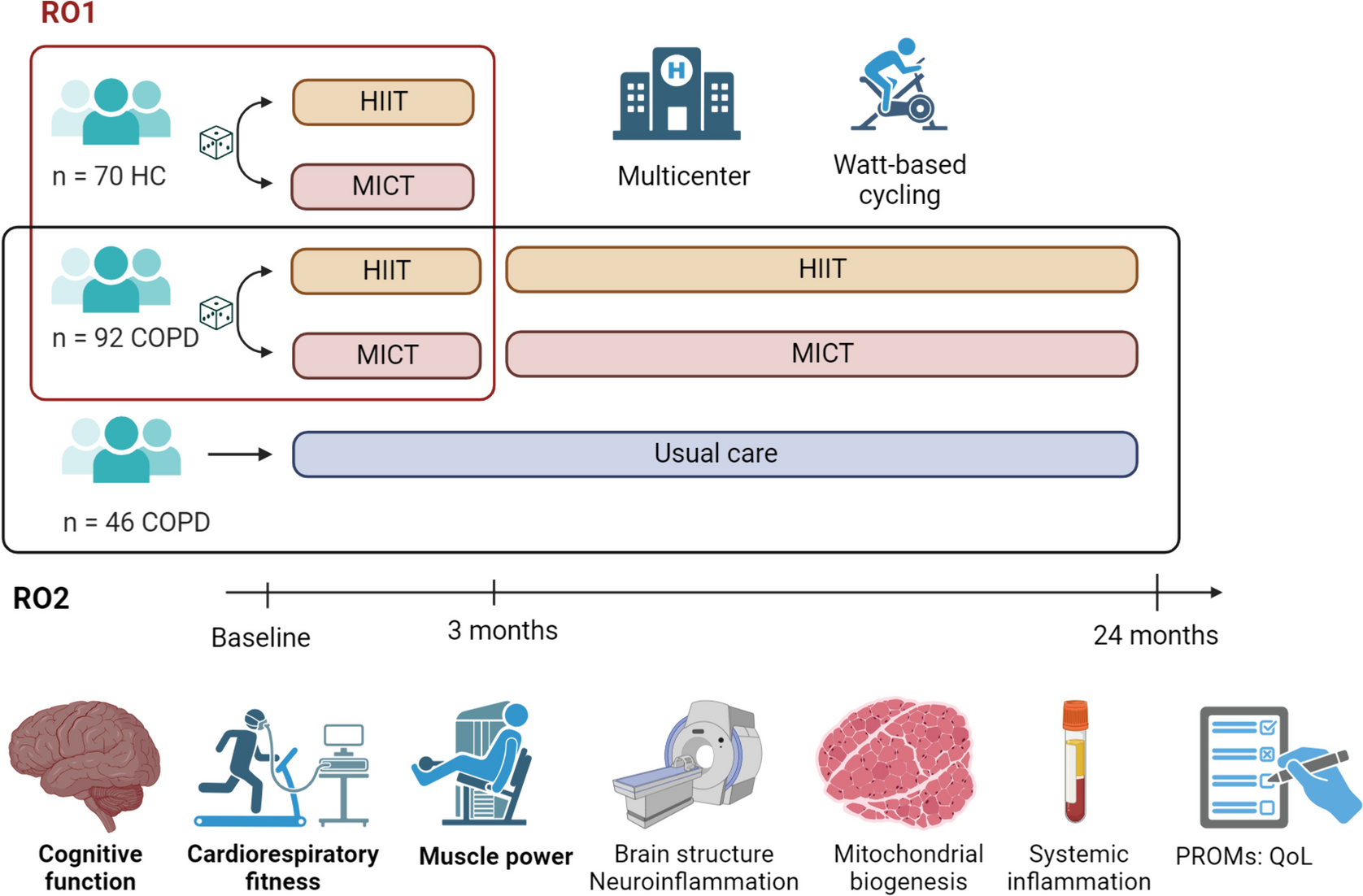


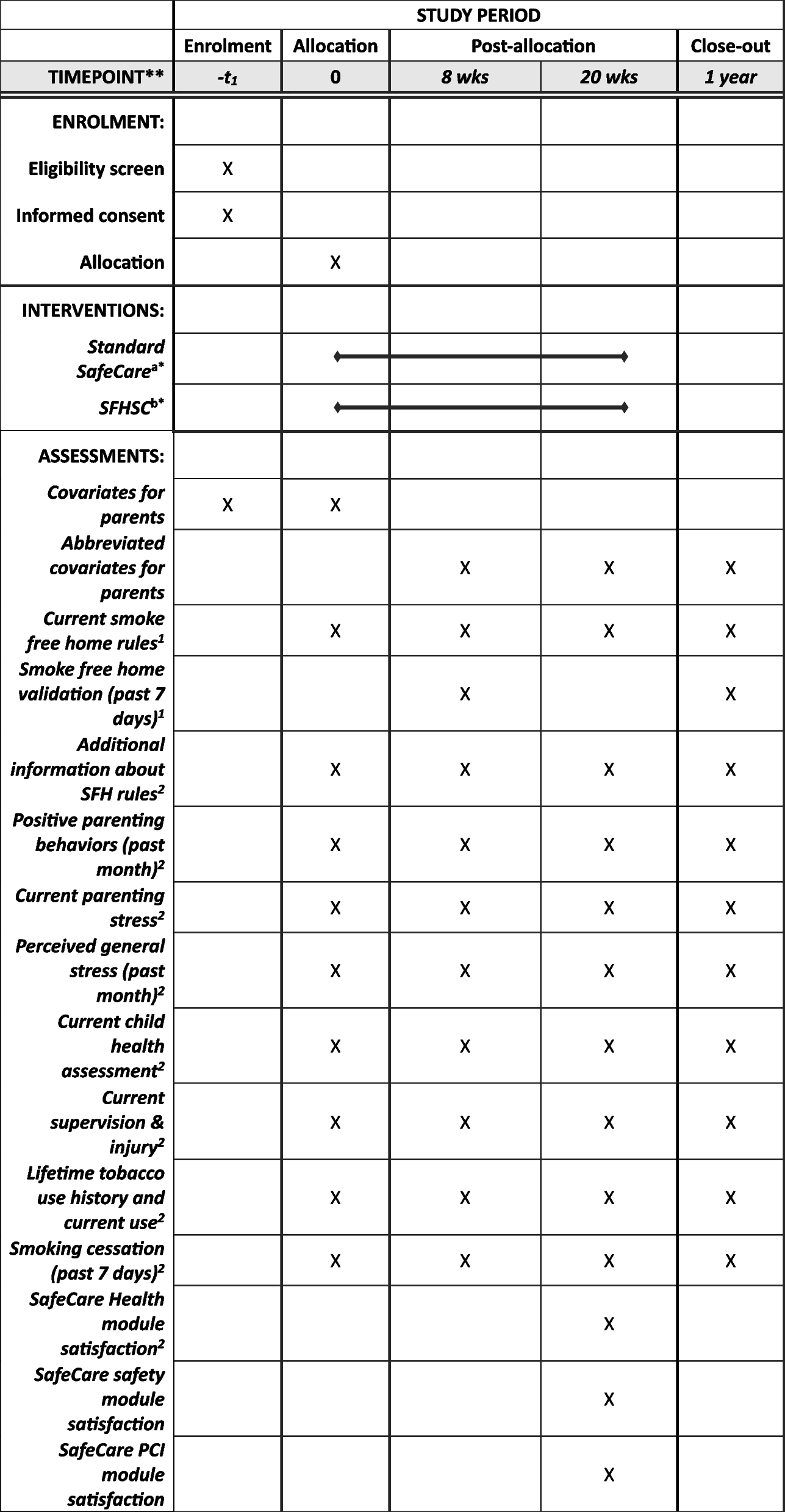
The study is a single-centre, open-label unblinded randomised controlled trial. The study is performed at Aarhus University Hospital in Denmark, which is a tertiary referral hospital that performs surgery for oesophageal cancer as a highly specialised function. All patients with oesophageal cancer in the form of adeno- or squamous cell carcinoma scheduled for surgery at Aarhus University Hospital are screened for study eligibility (Figs. 1 and 2).
Fig. 1
Standard Protocol Items: Recommendation for Interventional Trials (SPIRIT) figure. Schedule of enrolment, interventions and assessments for trial participants. For details of the inclusion and exclusion criteria, please refer to the applicable sections in the manuscript. For a detailed list of analyses performed on blood samples, please refer to Table 1. Abbreviations: F1 + 2, prothrombin factor 1 + 2; FU, follow-up; OP, operation
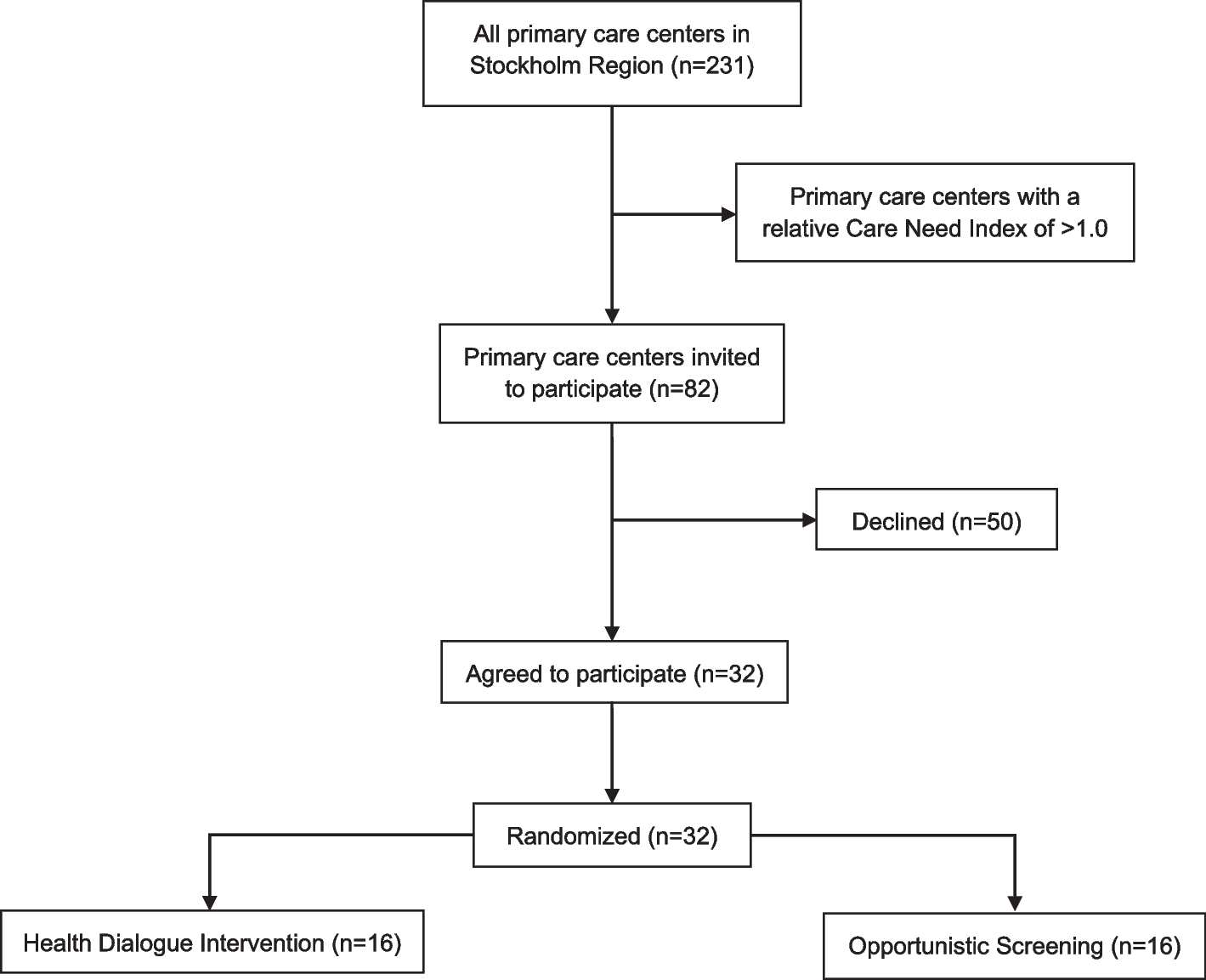
Fig. 2
Patient flow. Sample tubes and ultrasound probes mark the timing of blood sampling and ultrasound scanning, respectively. Patients in the standard and prolonged intervention group receive the same daily dose of 5000 international units (IU) with low molecular weight heparin (LMWH)
TimeframePatient inclusion started September 2021. We aim to include 100 patients, equally distributed between the intervention and the standard group.
Hypotheses 1.The intervention group of oesophageal cancer patients, who receive prolonged thromboprophylaxis with dalteparin has a lower VTE risk, expressed by a lower F1 + 2, 30 days after surgery than the standard group.
2.The intervention group does not demonstrate an increased bleeding tendency compared with the standard group.
The primary endpoint is the difference in F1 + 2 30 days after surgery between the intervention and the standard group.
The secondary endpoints are incidence of bleeding, VTE and mortality 30 days and 1 year after surgery.
Inclusion criteriaThe inclusion criteria are as follows:
1.Cancer located in oesophagus and/or cardia
2.Candidate for intended curative surgery
3.Age > 18 years
Exclusion criteriaThe study has the following exclusion criteria:
1.Known inherited bleeding disorder
2.Unable to provide informed consent
3.Arterial or venous thromboembolic events within the last 3 months
4.On-going anticoagulant treatment (vitamin K antagonists or direct oral anticoagulants)
5.Pregnant or has given birth within the last 3 months
6.Known allergy to the trial drug dalteparin
Inclusion and randomisationThe patients are screened and included in the study, after giving their informed consent, at their pre-operative interview at Aarhus University Hospital by study investigators or trained study nurses. The interview takes place approximately 1 week before surgery. After inclusion, the patients are randomised to receive either prolonged (30 days) or standard prophylaxis (approx. 10 days, prophylaxis is given until patient is discharged) with 5000 international units (IU) of the LMWH drug dalteparin daily. The randomisation sequence is generated with a 1:1 allocation using varying block sizes of 2, 4, 6 and 8 in the secure eCRF programme REDCap (Research Electronic Data Capture) (REDCap Consortium, Vanderbilt University Medical Center, Tennessee, USA), which is hosted by Aarhus University, Aarhus, Denmark.
BlindingThe patients and the responsible health care staff are not blinded to the intervention, as placebo injections are not utilised in either group. Injection of LMWHs often create a small haematoma that a placebo injection does not, and blinding would therefore be compromised. However, this does not affect the primary endpoint as it is purely biochemical. All laboratory analyses are performed blinded to the intervention and outcome.
InterventionThe formulation used in the study comes in pre-filled syringes containing 5000 IU, which is administered subcutaneously [23].
Patients in the standard group receive dalteparin until they are mobilised and discharged (approx. 10 days) and thus receive identical prophylaxis to patients not included in the study. The intervention group receives dalteparin for 30 days. Patients in the intervention group are taught to administer the drug themselves by nurses at the department prior to discharge or, if unable or unwilling to self-administer, the drug is administered by home nursing. Guidelines recommend 4 weeks of thromboprophylaxis with 5000 IE LMWH daily after laparoscopic and open abdominal gastrointestinal cancer surgery, and the dosage and administration form for the intervention group is therefore based on the current recommendations [6, 24].
The most common serious side effect of dalteparin is bleeding. However, there is a predictable dose–response effect, and for this reason, the drug traditionally does not require monitoring [25]. However, all patients included in the study will be monitored biochemically with platelet counts after surgery, which adds to the safety of the study and is to the benefit of the included patients in both standard treatment and intervention group. For a detailed list of side effects, please refer to the published product resumé [23].
To ensure compliance, patients are asked to keep an injection diary and return the empty syringes at the final outpatient control appointment. Other non-study pharmacological treatment will continue at the discretion of the physician responsible for the treatment of the patient.
Patient flowFigure 2 shows the inclusion and data collection flow from screening to the 30-day follow up. Furthermore, a follow-up review of patient records is performed 1 year after surgery (Fig. 1).
Data collectionClinical dataThe following information is recorded from electronic patient records and laboratory database.
At study entry:
1.Sex
2.Date of birth
3.Date of diagnosis
4.Medical history including tumour pathology
5.Latest blood electrolyte status, kidney function, and infection parameters
6.Eligibility criteria (i.e. all inclusion and exclusion criteria)
7.Medication
8.Preoperative Caprini score
At 30-day and 1-year follow-up:
1.Medical history including tumour pathology
2.Thromboembolic events including diagnosis by imaging
3.Major bleeding events, defined as leading to transfusion of 2 or more units of blood or packed red cells, decrease in haemoglobin of 2 g/dL, bleeding in critical sites, defined as spinal, epidural, intraocular, intracranial, pericardial, retroperitoneal or leading to death.
4.Medication
Furthermore, information from the registers is used to assign a World Health Organization (WHO) performance status at study entry and at each follow-up for each patient.
Blood sampling and analysesSamples are drawn from an arterial line if present. Otherwise, they are drawn from a peripheral vein applying minimal stasis. Samples are analysed immediately or frozen at − 80° C for batch analysis as appropriate. Table 1 shows an overview of the analyses performed on the collected samples.
Table 1 Overview of performed analysesUltrasound examinationsUltrasound scans are performed with Hitachi Aloka Arietta 850, GE healthcare LOGIQ E10 and E9 with linear transducer 3–7 MHz and L2–9 MHz. Examinations are performed by a radiologist before surgery and 30 days after surgery. The scan is performed on the patients’ lower extremities and include femoral communal and superficial veins, popliteal veins, venae saphena parva and magna as well as superficial and muscular veins and any symptomatic sites. If thrombosis is diagnosed on the preoperative scan, the patient is excluded from the study to receive a therapeutic dalteparin dosage.
Sample sizeDue to the rarity of oesophageal cancer, it is necessary to use surrogate biochemical markers for VTE as the primary endpoint. We have chosen F1 + 2 as the basis for the sample size calculation. Our research group has demonstrated this marker to be significantly elevated in patients with localised cancer [16]. Data on patients with oesophageal cancer do not exist yet; thus, the sample size calculation data are obtained from a group of patients with head and neck cancer, as this subset of patients have a VTE frequency close to that of patients with gastrointestinal cancer [3, 26]. F1 + 2 mean was 329 pmol/l with a standard deviation (SD) of 159 pmol/l. We considered a minimal relevant difference (MIREDIF) of F1 + 2 at 105 pmol/l between the two groups 30 days after surgery to be clinically relevant. Using a 5% significance level (2α) and a power of 90% (1-β), a minimum of 49 patients must be included in each group. To take missing data into consideration we plan to include 50 patients in each group.
Statistical analysesFor descriptive statistics, mean and SD will be calculated for data following a Gaussian distribution, and for data not following a Gaussian distribution, median and interquartile range (IQR) will be used.
Primary endpointThe difference in F1 + 2 between the two groups 30 days after surgery will be tested by an unpaired t-test if data follows a Gaussian distribution and by a Mann–Whitney U test if not.
Secondary endpointsIncidence of VTE and mortality 30 days as well as one year after surgery will be analysed unadjusted by the Kaplan–Meier method. The difference across sample time points will be investigated using repeated measurements-tests (mixed model analysis). Correlation tests will be performed using a Spearman test. A power calculation of the primary endpoint results will be performed if the expected number of patients are not included to assess the attained power of the study.
Risks and adverse eventsBlood samplingThere is a minor risk of infection, superficial thrombophlebitis and localised haematoma associated with blood sampling.
Ultrasound examinationUltrasound examination is painless, safe and with no radiation.
Dalteparin administrationAs described under the “ Intervention” section, dalteparin is primarily associated with an increased bleeding risk. All expected and unexpected adverse events and reactions occurring during study treatment will be documented in the applicable case report form (CRF) section in REDCap.
Serious adverse eventsA serious adverse event or reaction (SAE) is any untoward medical occurrence that at any dose results in death, is life threatening, requires inpatient hospitalisation or prolongation of existing hospitalisation or results in persistent or significant disability [27]. Thus, any death, whether associated with side effects of the study treatment or due to progressive disease, surgical complications or other causes are considered a SAE. In this study, the following are not considered a reportable SAE:
During the treatment period, all SAEs are documented on the SAE report form, and the sponsor is notified automatically. Based on the SAE report form, the sponsor-investigator and her delegates complete a SAE assessment sheet. These assessments are used for monitoring SAE survival and safety of the experimental treatment. Patients who withdraw from the study due to an adverse event or SAE are followed up to 30 days after the event.
Suspected unexpected serious adverse reactionsSuspected unexpected serious adverse reactions (SUSARs) are SAEs, which are:
1.Related to the study drug (dalteparin) and
2.Of a nature or severity that is not consistent with information in the reference document (the published product resume [23]). The sponsor will submit all available information about at SUSAR immediately and at the latest within 7 days after the event is known to the sponsor. The sponsor is responsible for informing the ethics committee, the regulatory authorities, the Danish Medicines Agency and all sub-investigators.
Furthermore, a safety report as specified by the Danish Medicines Agency is submitted to the authorities annually.
留言 (0)