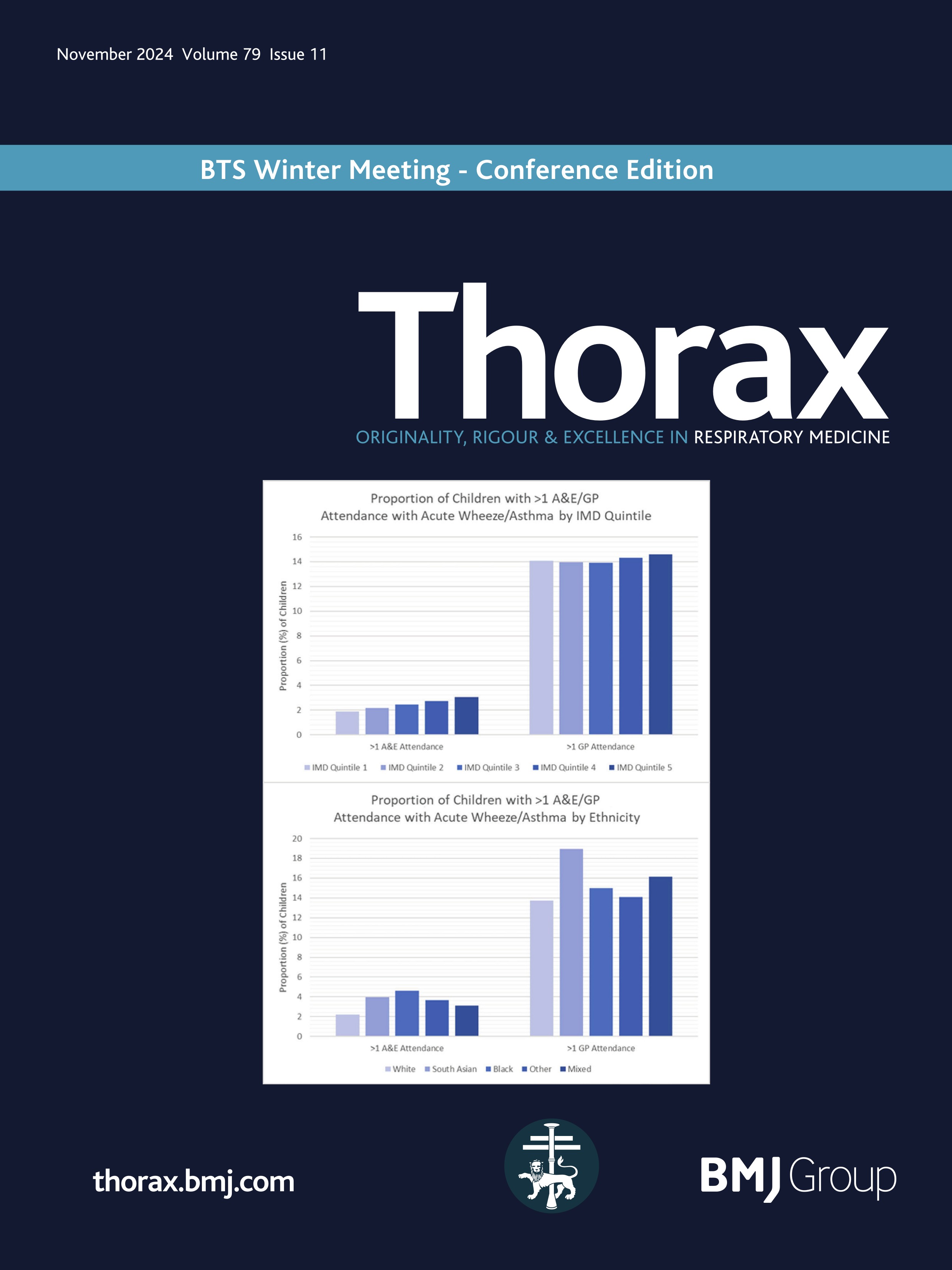
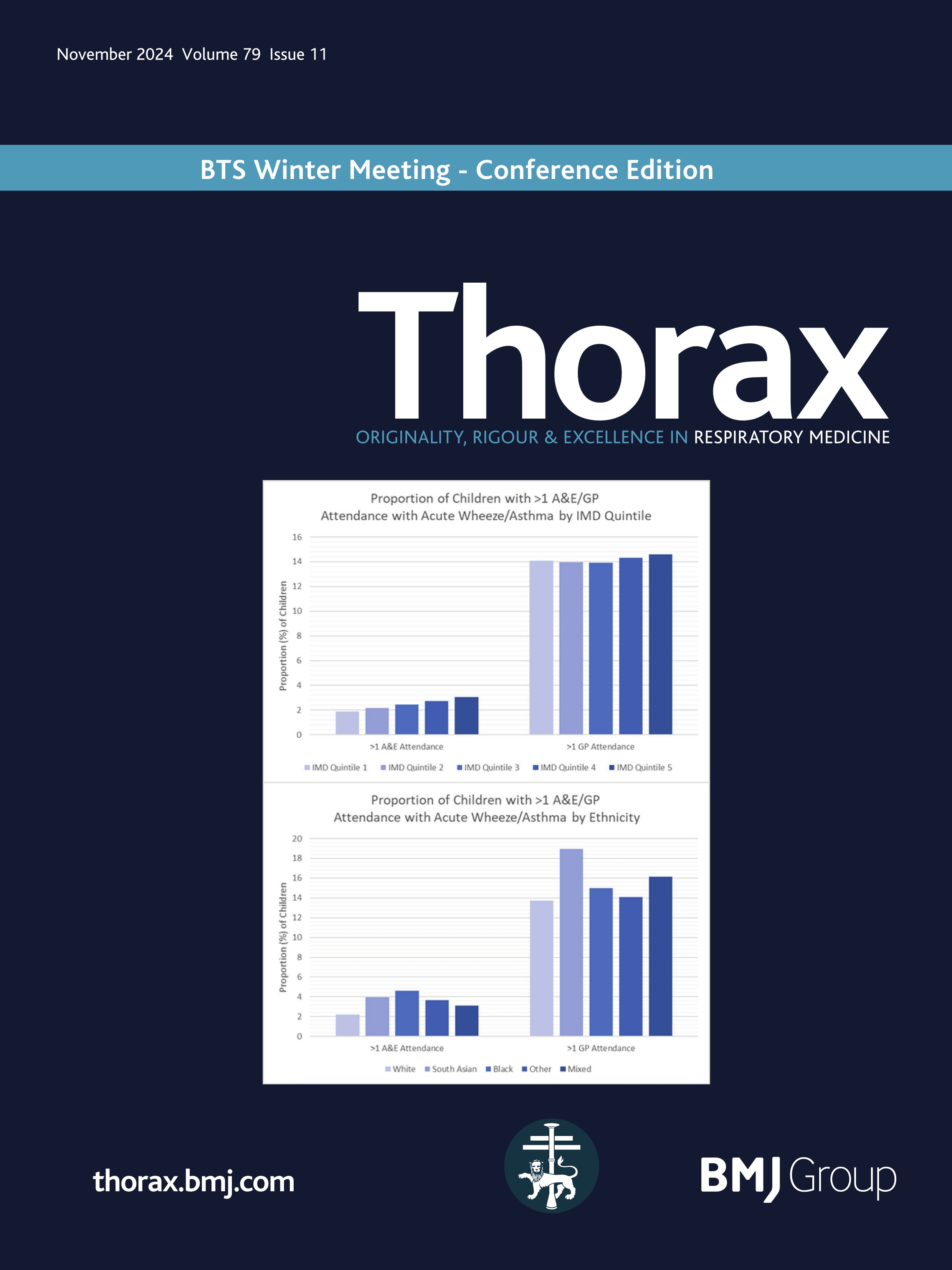
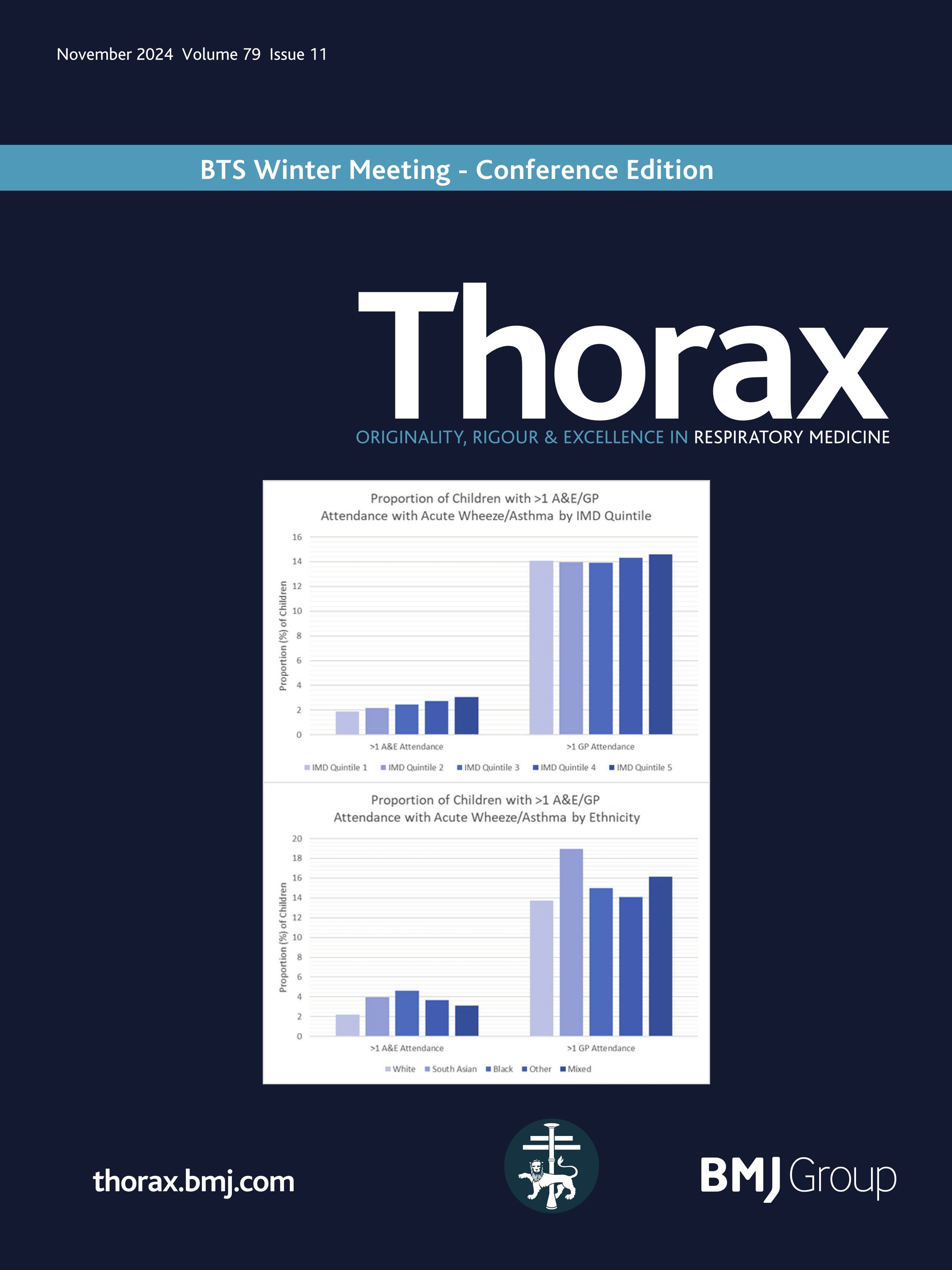
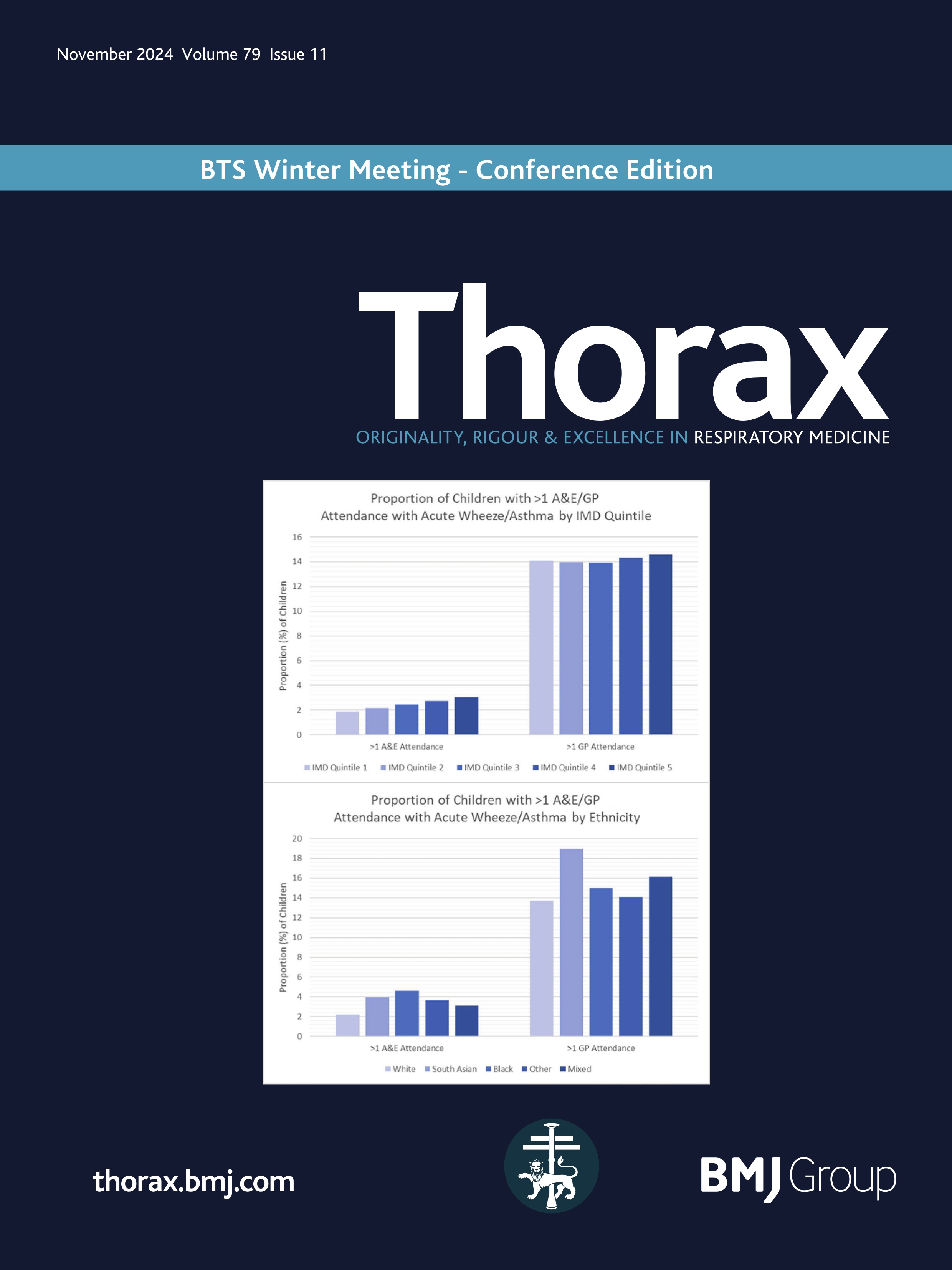
Rectal organoid morphometric analysis (ROMA)--re: optimising measurements automatically
Quantification of the physiological changes in people with cystic fibrosis (pwCF) has long been important for clinical diagnosis. Since the original discovery of abnormal sweat electrolytes in pwCF, the standardised pilocarpine iontophoresis sweat test was developed1 to clearly distinguish those with CF. While the sweat chloride test remains an important early step in the diagnostic algorithm, complementary testing methods are required to quantify the function of the cystic fibrosis transmembrane conductance regulator (CFTR) protein for both diagnosis and the development of new therapies.2
Building on this foundation, standardised protocols for the in vivo measurement of the nasal potential difference (NPD)3 were developed to evaluate sodium and chloride transport defects in the respiratory epithelium. This test played a pivotal role in the London gene therapy trial.4 5 However, the complexity of measuring CFTR function in respiratory tissues in vivo, particularly with interactions among multiple ion channels, underscored the need for more direct measurement techniques in vitro. The intestinal current measurement technique was developed, using rectal biopsies mounted in Ussing chambers to measure electrophysiological properties. However, Ussing chamber techniques requiring viable, functionally intact tissues presented significant challenges and limited widespread use.
To address this gap, the Beckman team cultured the rectal mucosal cells into miniature ‘organoids’ …

留言 (0)