
記住我
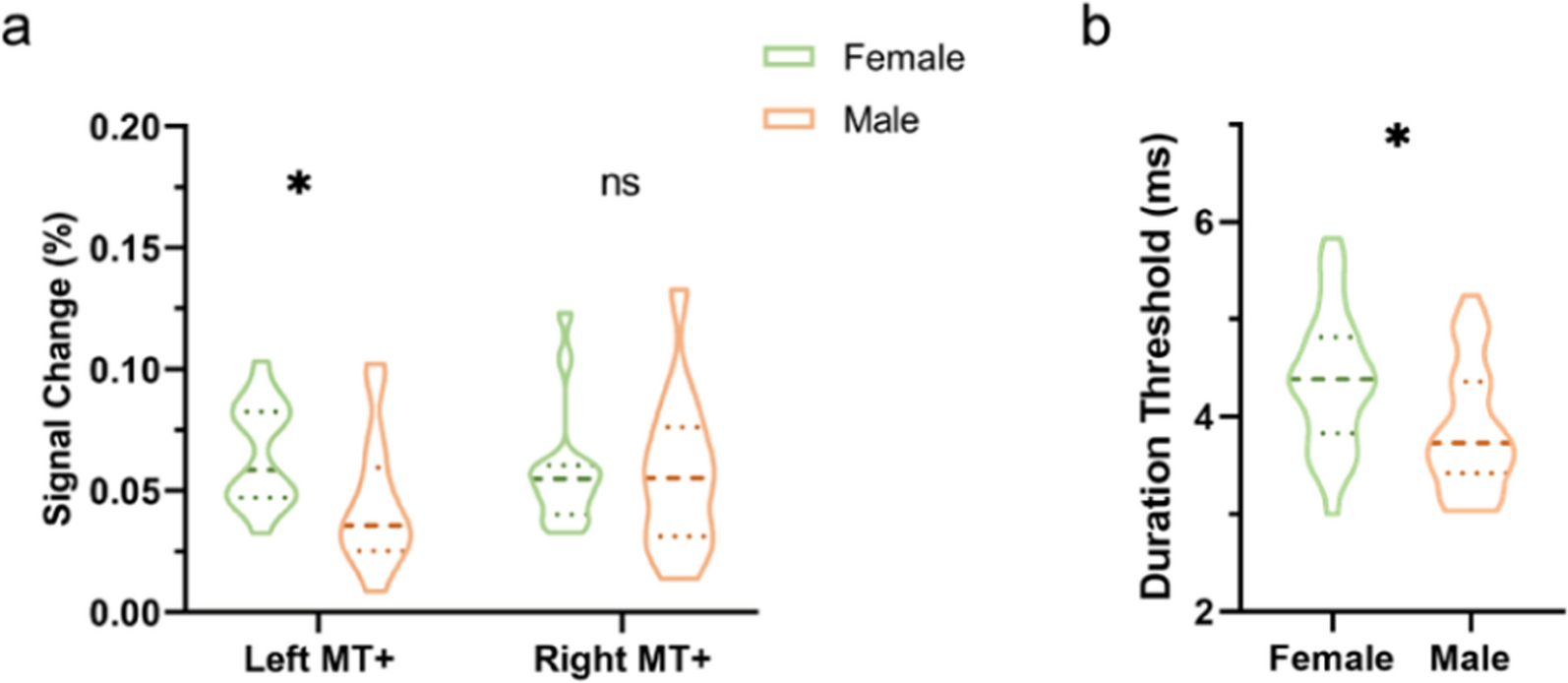

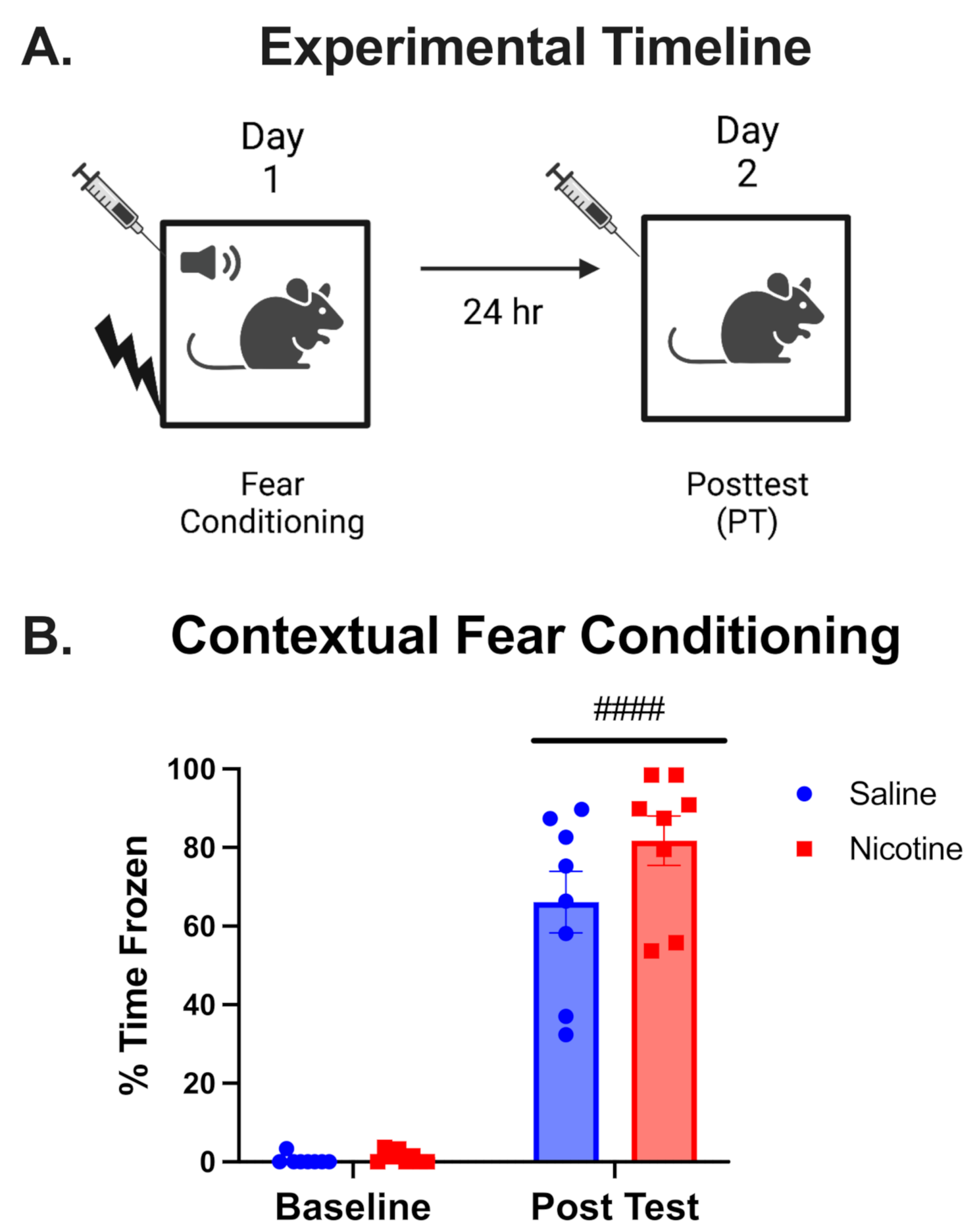
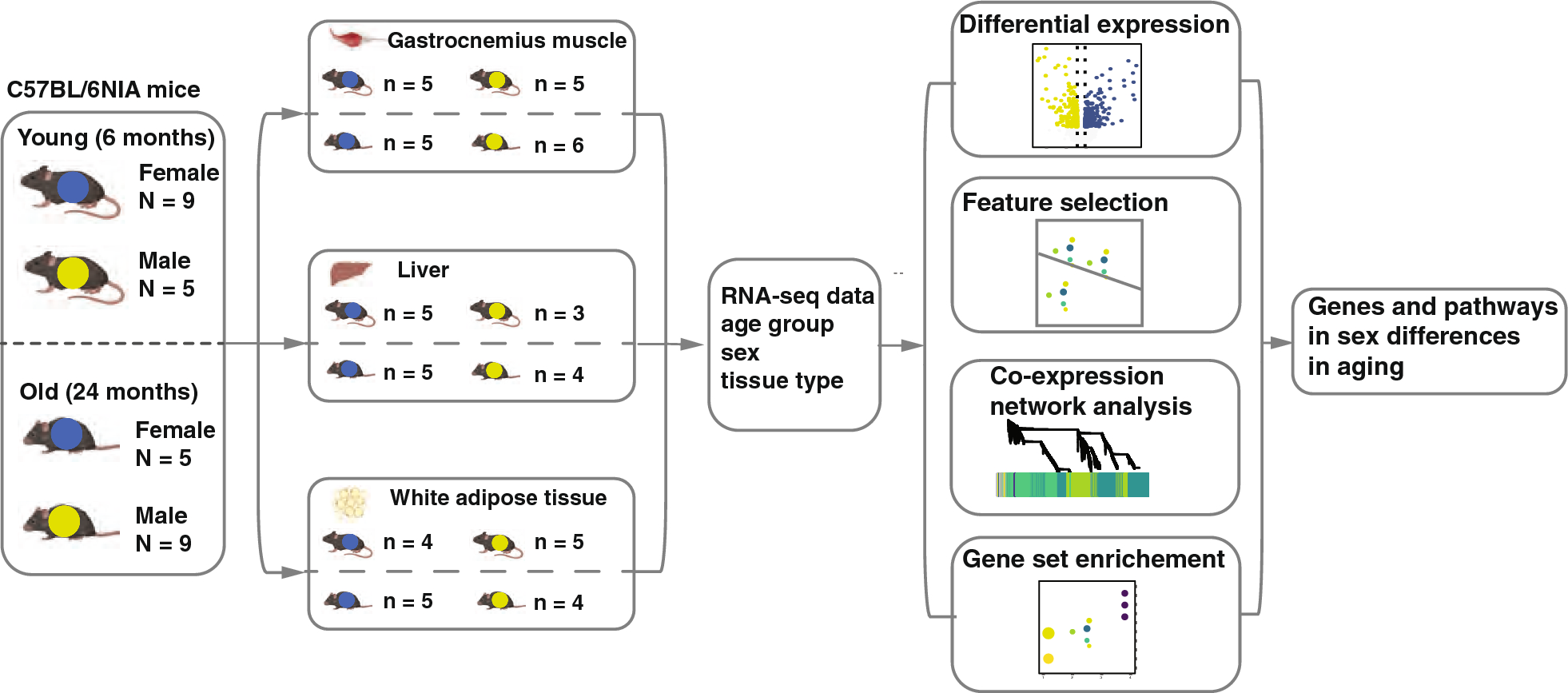
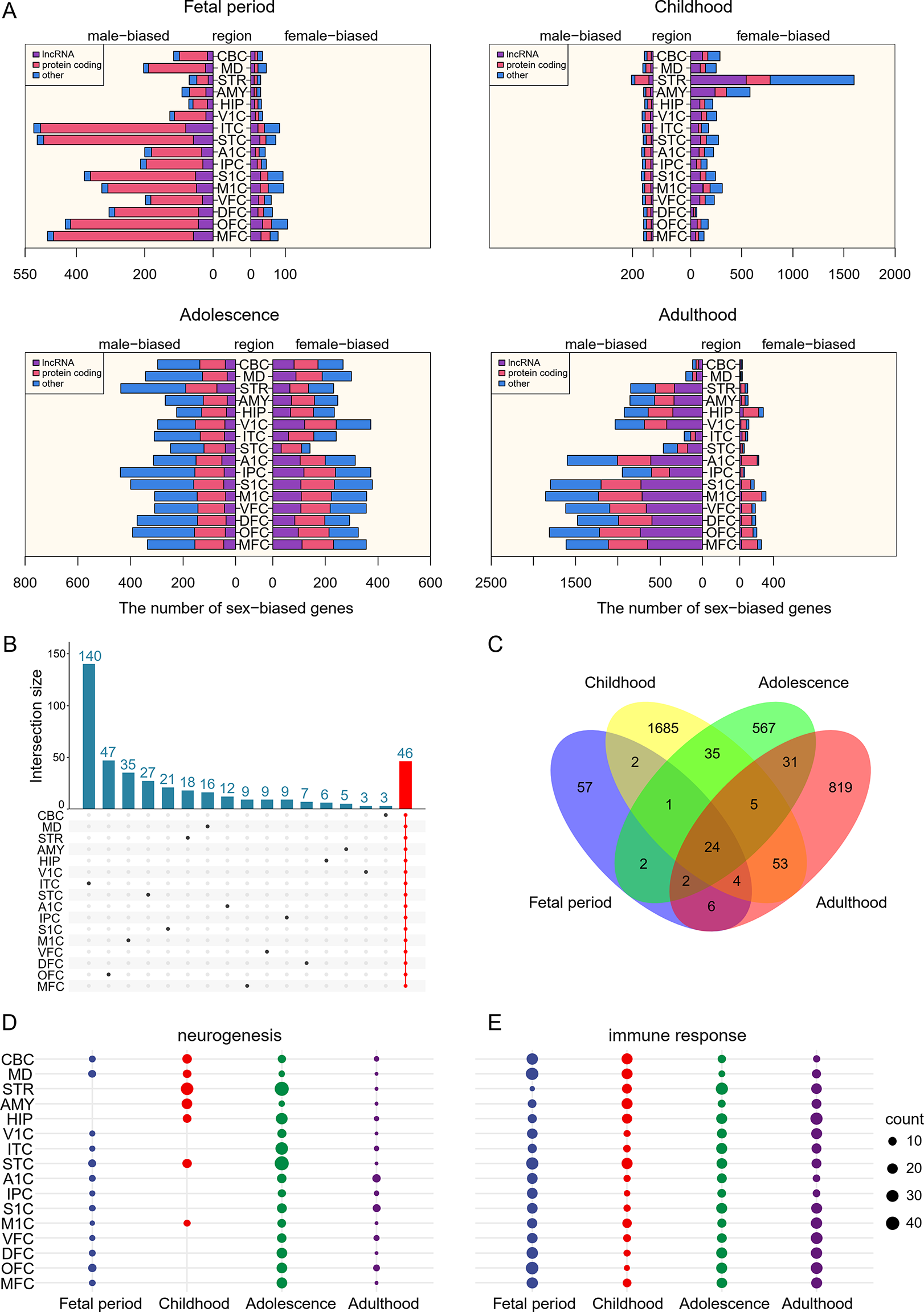





To understand the landscape of sex differences between males and females, we performed multiple systematic and bioinformatic analyses, such as sex-biased expression and sex-specific differential expression. From our analyses, we defined 4,328 genes with sex-biased signatures, including 298 genes with sex-biased mutations, 1,352 genes with highly significant sex-biased CpG sites (|difference beta value| >0.3), 2,114 sex-biased mRNAs, and 115 genes with sex-biased exon skipping which affect the open reading frame, and 461 genes with sex-biased RNA editing affect coding regions (Fig. 2A, Supplementary Table 2). We performed functional enrichment analysis on 4,328 genes and identified several themes among the sex-affected pathways. The first group related to cancer and oncogenic signaling pathways, including multiple cancer types. The second group related to cancer immune response includes the chemokine signaling pathway, cytokine-cytokine receptor interaction, viral protein interaction with cytokine, and cytokine receptors. The third group was the cell proliferation-related pathway including growth-factor signaling, cell proliferation pathways such as focal adhesion, cell adhesion molecules, NF-kappa B signaling pathway, PI3K-Akt signaling pathway, and central carbon metabolism in cancer (Fig. 2B).
Fig. 2
Sex-biased molecular signatures in human cancers. (A)The number of sex-biased signatures and number of sex-biased genes which are drug target. (B)Top 20 enriched KEGG pathways of genes which have at least one sex-biased signature. (C)From left to right are the expression of S100A9 in four cancers, and pink represents the female tumor patients, blue represents the male tumor patients; the expression of S100A9 five cancers, and pink represents the female tumor patients, black represents the female normal tissues. the drug and disease information of S100A9. Calcium has been approved by the FDA for the treatment of colon and rectal cancers(*p.adjusted < 0.05, **p.adjusted < 0.01, ***p.adjusted < 0.001, ****p.adjusted < 0.001)
In this work, we identified significant differences in gene expression between male and female tumor tissues across multiple cancer types. Specifically, 2114 genes exhibited differential expression between male and female tumors, with 1593 genes showing tumor-specific patterns and 521 genes displaying consistent differential expression across several cancer types. Furthermore, we observed 6,979 genes with distinct expression profiles between female tumors and normal female tissue across all cancers studied. Among these, the number of genes showing female-specific differential expression varied widely across cancer types, ranging from 203 to 2,924. Similarly, between male tumors and male normal tissues, 298 to 4204 genes showed differential expression across the 13 cancer types analyzed (Supplementary Fig. S4). Take S100A9 as an example, S100A9 was the target of five drugs, of which the FDA has approved calcium for the treatment of colon and rectal cancers [36]. S100A9 has been reported to regulate the behavior of cancer cells by inducing pre-metastatic cascades associated with cancer spread [37]. A previous study showed that S100a9 protected male lupus-prone NZBWF1 mice from disease development [38]. Our study found that S100A9 was differentially expressed between male and female patients in four cancer types (BRCA, DLBC, KIRP, SARC). Furthermore, through differential analysis between male/female tumors and their matched normal counterparts, we found that the expression difference of S100A9 between tumors and normal counterparts was also significantly different in the male and female groups (Fig. 2C). These results provide an overview of molecular differences between male and female cancer patients and imply sexual differentiation affects cancer incidence and progression.
Sex-biased somatic mutation data and single nucleotide polymorphisms showed different regulatory patterns between females and malesEvaluating genetic variants based on sex can reveal important sex disparities. Sex-biased effects of variants related to cell cycle and apoptosis have been described for many cancer types. From our analysis, a total of 298 genes in 18 cancers show the differential frequency between male and female tumor patients. Functional enrichment analysis found only 10 cancers sex-biased genes enriched in cancer related pathways. For example, 10 mutation gene showed sex-biased frequency in LIHC. Among them, CTNNB1, OBSCN, TP53, ALB and HERC2 show higher mutation frequency in male patients. BAP1, PCLO, TENM1, PIK3CA, PIKCA, and PKHD1L1 show higher mutation frequency in female patients. These genes enriched in hepatocellular carcinoma, central carbon metabolism in cancer, cellular senescence and lipid and atherosclerosis (Fig. 3A-B, Supplementary Fig. S1A). By integrating with six-biased gene expression, 3 genes with sex-biased mutation frequency also showed differential expression between male and female patients. Specifically, a previous study report CACNA1D overexpression and activate voltage-gated calcium channels in prostate cancer during androgen deprivation [39]. CACNA1D has been identified as a potential therapeutic target. It is upregulated in pancreatic cancer cells and associated with tumor growth and invasion. In this work, CACNA1D was found to show high mutation frequency in male SARC patients with a down-regulation transcript level. Recognizing and addressing sex differences patterns is essential for delivering personalized and effective patient care (Fig. 3C, Supplementary Fig. S5B).
Fig. 3
Sex-biased mutations(A) The number of gene with sex-biased mutation.(B) The functional enrichmentment result of gene with sex-biased mutation for each cancer type.(C) The sex-biased cis-eQTL of rs12256605 and HTR7 in BLCA. (D) The sex-biased cis-sQTL of rs12256605 and exon_skip_499249 in THCA.
Furthermore, single nucleotide polymorphism is the simplest form of DNA variation among individuals [40]. They are responsible for the diversity of individuals, drug response, and complex and common diseases [41]. SNPs can affect alternative splicing by modifying the sequence-specific binding affinity of the splicing factors to the pre-mRNAs [42], resulting in splicing quantitative trait locus. And may influence promoter activity [40], resulting in the expression of quantitative trait locus. To explore the sex-biased effects of SNPs on gene expression and alternative splicing, we performed eQTL and sQTL analyses for the male and female patients, separately. 1,654,515 sex-biased cis-eQTLs pairs and 76,615 sex-biased cis-sQTL pairs were identified. Of 1,654,515 sex-biased cis-eQTLs, 214,335 were found to occur in 2,294 drug target genes including 254 cancer therapeutic targets (Supplementary Fig. S5C). Based on the position of the SNPs, we identified 578, 408, 163, 731, and 430 SNPs located in the promoter, enhancer, coding region (CDS), exon, and UTR of the gene, and 11,055 SNPs in the gene body region. For example, HTR7 promoted laryngeal cancer growth through the activation of the PI3K/AKT pathway [43]. Rs12256605(chr10:9086171, G to C) located in the promoter region of HTR7 showed a significant correlation with the expression of HTR7 only in male BLCA (Fig. 3D). HTR7 is the drug target of gilteritinib, the sex-based cis-eQTL regulation of HTR7 may lead to different treatment effects of gilteritinib and will help to design personalized medicine.
Sex-biased sQTL analysis identified 76,615 sex-biased cis-sQTL pairs including 72,349 SNPs and 2,872 ES events in 2,256 genes. Furthermore, we overlapped the genomic coordinates of exon skipping events and SNPs and identified 17 SNPs located in the skipping regions of 14 ES events showing the sex-biased cis-sQTL regulations. For example, SLC2A8 belongs to the solute carrier 2 A family [44]. The skipping of exon5 (exon_skip_499249, chr9:127402556–127402753) in SLC2A8 was affected by rs1138739 in THCA female group. The mutation of rs1138739 in the skipped exon region may lead to exon skipping in female patients, resulting in frameshift ORF that may affect the cancer progression (Fig. 3E).
DNA methylation-related sex-biased regulationsMethylation appears to influence gene expression and splicing patterns by affecting the interactions of DNA with both chromatin proteins and specific transcription factors [45, 46]. Combining the mRNA and DNA methylation analyses, we identified 1,672 sex-biased CpG sites in the gene promoter region of 582 sex-biased genes (Fig. 4A, Supplementary Table 3). For example, SRPX is a tumor-suppressor gene that was reported to be downregulated in a variety of human tumor cells and tissues. A previous study reported that Srpx-knockout mice generated various tumors, including lymphomas, lung cancer, and hepatomas [47, 48]. We found SRPX showed differential expression between male and female tumor patients. Our data suggested that total 10 CpG sites located in the SRPX promoter region showed higher methylation in female SARC patients inhibiting the expression of SRPX in female SARC patients (Fig. 4B, Supplementary Fig. 6A). This result is consistent with the established role of DNA methylation in gene regulation that hypermethylation leads to gene silencing [49].
Fig. 4
DNA methylation-related sex-biased regulations and Sex-biased RNA editing. (A)The number of sex-biased related DNA methylation mediated genes. (B) Promoter methylation affected SRPX expression in SARC. Left is the heatmap of 10 CpG sites in SRPX promoter regions. (C) CpG sites associated with S100A9 in KIRP. (D) The PSI values of exon_skip_499249 in low methylation (0 ~ 0.2), middle methylation (0.2 ~ 0.6), and high methylation (0.6 ~ 1) groups. The patients were categorized into three groups based on the beta value of cg24870846. (E) RNA editing frequence of chr11:61007595 in CD6. (F) chr12:68851361 A-to-I editing in CPM 3’UTR. From left to right are the RNA editing frequency of chr12:68851361; the expression of CPM; and the mechanism of miRNA binding increase in CPM 3’UTR region through RNA editing variant(*FDR < 0.05, ** FDR < 0.01, ***FDR < 0.001, ****FDR < 0.0001).
To further analyze the effect of sex-biased methylation site, we integrated multi-omics and performed eQTM and sQTM analyses for the male and female patients. Our analyses identified 764,720 sex-biased cis-eQTM and 9,442 sex-biased cis-sQTM. 1,305 sex-biased cis-eQTMs were found in 338 drug target genes, including 38 cancer therapeutic target genes (Supplementary Fig. 5C). Detailly, three CpG sites in CpG island (chr1:153260996–153261837) were associated with S100A9 expression in the KIRP female group. The high methylation in the promoter region inhibits the expression of S100A9, which may influence S100A9-targeted drug resistance and cancer progression in female patients (Fig. 4C, Supplementary Fig. S6B).
Methylation at the DNA level can modulate the elongation rate of RNA polymerase II or the format of a protein bridge of splicing factor to affect the exon skipping [50]. This work identified 9,442 sex-biased cis-sQTMs, including 8,738 CpG sites and 2,008 ES events. Among them, 27 exon-skipping events occurred in the cancer therapeutic target genes. For example, TNK2 was the target of entrectinib [33], which has been approved to treat non-small cell lung cancer and solid tumors in the body [51, 52]. Our results showed the inclusion of exon 14 in TNK2 (exon_skip_420474, chr3:195867168–195867258) was positively correlated with cg24870846 in male LGG patients (Fig. 4D). Low methylation of cg24870846 could cause the in-frame ORF and result in the loss of the ‘UBA domain’, and ‘Activated CDC42 kinase 1 chain’ of TNK2. Overall, SexAnnoDB provides epigenetic factors that potentially influence gene expression and splicing. These factors may affect the downstream regulations of cancer progression. All filtered sex-biased cis-eQTMs and sex-biased cis-sQTMs were shown in SexAnnoDB.
Sex-biased RNA editing affected the protein function and gene expressionRNA editing events in coding regions can alter amino acid sequences and have a chance to affect protein functions. To study this, we first identified 19,503 RNA editing site show difference frequency between male and female tumor patients. 14 of them in coding region might cause the non-synonymous SNV of 12 genes in 9 cancer types. Specifically, CD6 is a cell surface glycoprotein on human lymphocytes, including T cells and natural killer (NK) cells. recent studies strongly suggest that anti-CD6 should also be evaluated as a new cancer immunotherapy [53,54,55]. In this work, we found an RNA editing events in the position of chr11:61007595 show the differential frequency between male and female patients in two cancer types (SKCM, STAD). The RNA editing leads to the S/G changes of SRCRdomain in the CD6 protein (Fig. 4E, Supplementary Fig. S6C).
Combining the mRNA and RNA editing analyses, we identified 22 sex-biased RNA editing sites of 14 sex-biased genes(Supplementary Table3). Among them, 6 RNA editing sites were in 5 gene 3’UTR region. From the analysis of A-to-I RNA editing effects on miRNA regulation in CAeditome, 3 RNA editing will cause mRNA to gain miRNA binding sites, and 1 RNA editing will cause mRNA to lose miRNA binding sites (Supplementary Table S1). CPM can significantly inhibit cell viability, ROS production, intracellular pH, migration in hypoxic lung cancer cells, and angiogenesis of HUVECs under hypoxia through the inhibition of APEX1/HIF-1α interaction [56]. From our analysis, one RNA editing site in the CPM 3’UTR region shows higher editing frequency in SARC female patients. The editing alternation would cause the gain of four miRNAs binding and lead to downregulating its expression (Fig. 4F). Overall, the sex-biased RNA editing event has the potential to lead to protein loss of function or affect gene expression, which shows it’s potential to be a therapy target for precision treatment.
RBP-ES regulatory networks identified the functional ES events related to cancer progressionRBPs are indeed primary regulators of splicing events. They interact with pre-mRNA molecules to facilitate or inhibit splicing, thereby influencing the production of mature mRNA transcripts [57]. PANDA is a method for estimating gene regulatory networks that were not specifically developed for RBP-ES networks, here we performed PANDA in 8 RBPs knockdown/out RNA-seq data to evaluate the performance of PANDA. We utilized RNA-seq and eCLIP data to identify the RBP-ES regulation pairs in RBP knocked. PANDA demonstrated moderate performance, achieving an AUC of 0.68 in identifying RBP-ES regulations (Supplementary Fig. 7, Supplementary method). Thus, in this work, we constructed RBP-ES networks using PANDA based on the RBP-ES interaction, expression profiles of RBP, PSI values of ES events, and protein-protein interactions to study the effects of RPB on ES events in differential gender groups.
Our analyses identified 56,185 sex-biased RBP-ES regulatory edges, including 18,790 ES events and 74 RBPs. Functional annotation of ES events showed that 13,132 targeting ES events could lead to open reading frame (ORF) alternation, of which 4,257 could lead to In-frame alternation of ORF and resulted in loss of protein function. Combined with drug information, we identified 2,192 targeting ES events appearing in drug target genes. 324 targeting ES events appeared in cancer therapeutic target genes (Fig. 5A and B). For example, MET lost the binding site of E3 ubiquitin ligase CBL through an exon 14 skipping event, resulting in an increased expression level of MET. MET amplification drives the proliferation of tumor cells [58, 59]. From the drug and disease annotation, Multiple tyrosine kinase inhibitors, such as crizotinib, cabozantinib, and capmatinib have been used to treat patients with MET exon 14 skipping. In addition, crizotinib, cabozantinib, and capmatinib have been approved by FDA to treat non-small cell lung cancer (NSCLC), non-hodgkin lymphoma (NHL), renal cell cancer (RCC), liver cancer. Our result found exon14 skipping (exon_skip_470684, chr7:116774880–116775111) of the MET gene showed the male-based regulation by MSI1 in LIHC, UVM, and female-biased regulation by MSI1 in ESCA, KIRC, PAAD, THCA. These results will help to design a personalized medical plan for the patient with MET exon 14 skipping (Fig. 5C).
Fig. 5
Sex-biased RBP-ES regulatory network analysis. (A) The pipeline to identify the functional ES events in the sex-biased RBP-ES network. (B) Number of RBP targeting ES events in sex-biased RBP-ES network of individual cancer type. (C) The sex-biased MSI1-exon_skip_470684 edges include two male-biased edges (LIHC, UVM) and four female-biased edges (ESCA, KIRC, PAAD, THCA). (D) Left: The expression of RBM24 in LIHC. Right: RBM24-ES regulatory network in LIHC. (*p. adjusted < 0.05, **p.adjusted < 0.01, ***p.adjusted < 0.001, ****p.adjusted < 0.001)
For 72 RBPs related to sex-biased RBP-ES regulation, 3 RBPs were differentially expressed between male and female patients (Supplementary Table S6). For example, RBM24 was sex-biased expressed in LIHC and targeted 10 ES events, 3 of which were frame-shift ES events and 5 were in-frame ES events. Among them, exon3 of TXN was the target of phenethyl isothiocyanate. We found RBM24 also targeted TXN exon 3 skipping (exon_skip_505647, chr9:110244777–110244843) in CHOL, COAD, KIRC, KIRP, and PAAD female patients and PCPG and STAD male patients. The TXN gene encodes a protein involved in the process of the regulation of the cellular redox state [60]. In cancer cells, increased TXN expression increases proliferation and cell survival [61, 62]. Exon3 skipping of TXN can cause the in-frame alterations of TXN-202, leading to the loss function of “Beta strand”, “Chain”, “Disulfide bond”, “Domain”, “Helix”, “Modified residue”, “Mutagenesis”, and “Sequence conflict” to TXN protein(H9ZYJ2). Therefore, our results indicated that the downregulation of RBM24 in female LIHC patients could cause the loss of normal function of TXN and inhibit cancer progression (Fig. 5D).
TF-gene regulatory network reveals the sex-biased expression regulation between female and male patientsGene expression is controlled by complex networks of interacting factors. What’s more, the dysregulation of gene regulatory processes can lead to multiple diseases, including cancer. TF is one of the most important regulators of gene expression [63,64,65]. To explore the sex-biased TF-gene regulation, we constructed TF-gene regulatory networks based on TF-promoter interaction information, protein-protein interaction, and expression profiles for male and female patients, respectively (Fig. 6A). A total of 340,415 sex-biased edges were identified, including 169,132 male-biased edges and 171,283 female edges (Fig. 6B). 590 edges showed sex-biased regulation across more than 5 cancer types. Combined with differential analyses, we found that 243 targeting genes were differentially expressed between male and female patients. Besides, 71 sex-biased genes with sex-biased TF-gene regulations were also targeted by 491 drugs. 12 of the targeting sex-biased genes (S100A9, SLAMF7, TNFRSF17, CCND1, CD274, TOP2A, HTR2B, CD3D, SSTR2, ORM2, ORM1, TNF) have been reported previously as cancer therapeutic targets (Fig. 6C, Supplementary Fig. S8,Supplementary Table S7).
Fig. 6
Sex-biased TF-Gene regulatory network analysis. (A) The pipeline to identify the cancer therapeutic target genes in the sex-biased TF-gene network. (B) The number of sex-biased edges for each cancer type. (C) The number of targeting genes (white bar) and drug-targeted genes (red bar) in the sex-biased TF-gene network for each cancer type. (D) Left: the volcano plot of sex-biased genes in SARC. Pink marked as female-biased genes (log2FC< − 1 and p.adjusted < 0.05), blue marked as male-biased genes (log2FC > 1 and p.adjusted < 0.05). Right: sex-biased TF-Gene regulatory network of 7 sex-biased drug targeted genes in SARC
In SARC, a total of 289 targeting genes were reported as drug targets. Among them, SBK1 and CHRM2 were upregulated in male patients, and ADH1C, ADH1B, S100A9, CCL11, and KDM5D were upregulated in female groups. We found the female-biased genes owned more female-biased TF targeting edges and the male-biased genes owned more male-biased TF targeting edges. For example, S100A9 was differentially expressed between male and female SARC patients (Fig. 2C). Our data suggested that four TFs targeting S100A9 in the male group were responsible for the upregulation of S100A9 in male patients and affected the cancer progression (Fig. 6D). This is consistent with the fact that TF targeting the gene promoter will promote the gene expression [66]. The sex-biased TF-gene network will help us to understand the sex difference regulation of the sex-biased genes in cancer.
Sex-biased competitive endogenous RNA networkCompetitive endogenous RNAs have revealed a new mechanism of interactions among diverse types of RNAs and play crucial roles in multiple biological processes and the development of neoplasms [67]. We constructed sex-biased ceRNAs regulatory networks for sex-biased lncRNA, miRNA, and mRNA. Our analyses found that 25 male-biased mRNAs and 312 female-biased mRNAs were involved in ceRNA regulation. Based on the drug information, 29 sex-biased mRNAs were drug-targeted genes and six (CD38, PGR, ESR1, GABRP, F3, PRKCB) of them were targeted by cancer therapeutic drugs (Fig. 7A,Supplementary Table S8). For example, ESR1 is the target of raloxifene, toremifene, tamoxifen, and fulvestrant which are reported to prevent or treat breast cancer [33, 68,69,70,71] (Fig. 7B). ESR1 mutations are a common cause of acquired resistance to the backbone of therapy in the metastatic hormone receptor-positive breast cancer [72,73,74]. A previous study reported that the microRNA hsa-mir-206 decreased endogenous ERα mRNA and protein levels in human MCF-7 breast cancer cells by acting through two specific hsa-mir-206 target sites within the 3′-UTR of the human ERα transcript [75]. In our study, the hsa-mir-206 was predicted to bind to both 3′-UTRs of ESR1 and HAND2-AS1, indicating HAND2-AS1 protects ESR1 through competing for the miRNA binding sites of hsa-mir-206 in SARC female patients (Fig. 7C). In a word, our study identified potential sex-biased ceRNA regulation, providing insights for further research on the molecular mechanisms and potential prognosis biomarkers.
Fig. 7
Sex-biased ceRNAs in human cancers. (A) The pipeline to identify the cancer therapeutic targets in sex-biased ceRNA network. (B)Related drug information of ESR1. (C) The expression of HAND2-AS1(lncRNA), hsa-mir-206(miRNA), and ESR1(mRNA) in SARC (*p. adjusted < 0.05, **p.adjusted < 0.01, ***p.adjusted < 0.001, ****p.adjusted < 0.001)
Sex effects on cancer therapeutic target genesTo investigate the clinical implications of sex-biased molecular signatures, we focused on 2,526 therapeutic targets in DrugBank, including 205 cancer therapeutic targets of FDA-approved drugs. Across the various molecule dimensions we examined, we found that 131 drug target genes were associated with at least one type of sex-biased signature, and 126 were cancer therapeutic target genes (Supplementary Table S9). Among these genes, nine genes (CD274, CD38, CTLA4, EGFR, ERBB3, IL6, LCK, PDCD1, and RET) showed sex differences not only in mRNA expression but also in protein levels. These genes also showed sex-biased cis-eQTLs and cis-eQTMs regulation, indicating that sex differences at the transcriptome level are partially the result of corresponding sex-biased epigenetic and genetic regulations. Besides, four sex-biased exon skipping events in cancer therapeutic target genes (FDPS, FGFR1, CDK4, and IMPDH2) could cause the loss of partial protein function, which may contribute to a differential drug response rate in male and female patients (Fig. 8A). Moreover, we found six sex-biased genes (EGFR, CDK6, PRKCB, PDCD1, KCNH2, FCGR3B, and TOP2A) in BRCA were the therapeutic targets of breast cancer (Supplementary Fig. S9). EGFR is one of the first identified important targets of these novel antitumor agents [76]. Two drugs (Lapatinib and neratinib) for the treatment of breast cancer have been evaluated in several studies. Our result showed EGFR had differential expression both at the transcriptome level and proteome level, which are particularly important in determining sex-biased drug response rates (Fig. 8B-E). These results highlight the clinical importance of sex-biased molecular signatures.
Fig. 8
The sex-biased effect on cancer therapeutic targets. (A) Sex-biased cancer therapeutic drug target genes. The cancer-drug pairs were collected from the National Cancer Institute (https://www.cancer.gov/about-cancer/treatment/drugs/cancer-type) and the drug-gene pairs were collected from DrugBank. (B)The volcano plot of sex-biased genes in BRCA. Pink marked as female-biased genes (log2FC<–1 and p.adjusted < 0.05), blue marked as male-biased genes (log2FC > 1 and p.adjusted < 0.05). (C) The gene expression of EGFR in BRCA. (D) The protein expression of EGFR in BRCA. (E) Cancer-drug-sex-biased gene information in BRCA (*p. adjusted < 0.05, **p.adjusted < 0.01, ***p.adjusted < 0.001, ****p.adjusted < 0.001)
Furthermore, from a manual review of the abstracts, we found that there are 31 documentaries supporting 40 genes showing different patterns between females and males. Among them, 13 genes (AFP, ASNS, BEX4, BRAF, ESR1, FST, KIT, NR3C1, STAT3, TP53, TRPA1, AR, FST) were reported to show sex differences in cancer (Supplementary Table 10). Among them, ESR1 is involved in a sex-biased competitive endogenous RNA network, ESR1 competing for the miRNA binding sites of hsa-mir-206 in SARC female patients, TP53 shows higher mutation frequency in male LIHC patients, which is consistent with the previous research (Fig. 7, Supplementary Fig. S5A). These findings underscore the importance of considering sex-specific differences in cancer research, as they shed light on distinct genetic patterns and potential molecular mechanisms that may contribute to gender-specific variations in cancer development and progression.
留言 (0)