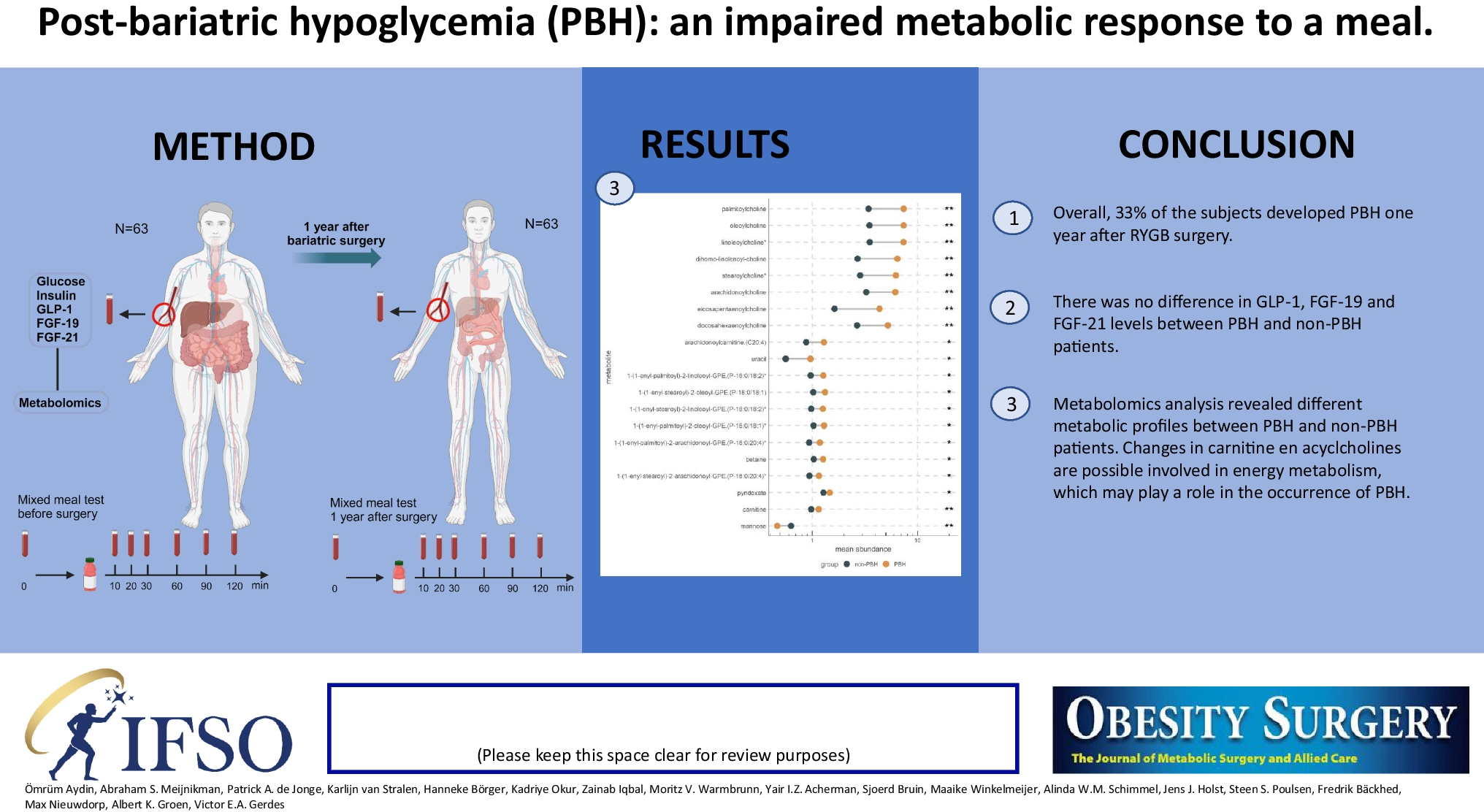
In our search for the culprits causing PBH, we measured an extended array of factors that have been suggested to play a role in induction of hypoglycemia after bariatric surgery. We could not confirm a direct role for GLP-1, which was reported in other studies [6, 10]. However, by carrying out non-targeted plasma metabolomics, we did obtain an indication for an interesting novel pathway. We observed increased levels of acylcholines that may signal inhibition of mitochondrial fatty acid beta-oxidation inducing stimulation of glycolysis thus lowering plasma glucose levels. Although our findings are preliminary and need reconfirmation in larger cohorts, we propose that acylcholines might be players in the development of PBH.
The anatomical changes induced by RYGB surgery are known to cause an increased glucose uptake in the first 30 min after a meal [22]. We confirmed a pronounced insulin and GLP-1 response 1 year after surgery, but for the latter there was no difference between the PBH and non-PBH groups. Insulin production did significantly differ between the two groups (p < 0.05), but the differences are mainly in the first 30 min after ingestion of a meal. Interestingly, at 120 min after initiating the MMT, insulin levels were identical in PBH and non-PBH groups. In addition, patients with and without PBH were characterized by similar GLP1, FGF-19, and FGF-21 plasma levels and the same amount of enteroendocrine L-cell density in the jejunum. Also, our observed prevalence of PBH of 33.3% is in line with other reports [2]. Previous studies suggested this to be a result of pancreatic beta-cell hyperplasia and hypertrophy, possibly as a result of the increased GLP-1 levels [4, 23]. However, although our results indicate that GLP1 contributes to the changed pattern after surgery, this incretin does not seem to explain the exaggerated response in our patients with PBH [24]. Also, post-mortem analysis did not show any pancreatic beta-cell hyperplasia after bariatric surgery [25]. Finally, although it was suggested that insulin resistance might be protective against PBH [26], we did not observe differences in HOMA-IR between the PBH and non-PBH groups.
In line with previous studies, we also confirmed that stimulated GLP-1 release was increased after surgery [9]. In this regard, it has been suggested that proliferation of jejunal enteroendocrine L cells could contribute to this response [27]. However, in our study, L-cell density at time of surgery did not show any correlation with GLP-1 or PBH.
Other researchers reported a possible non-insulin dependent role for FGF-19 in developing PBH, although the mechanism was unclear [12]. In our study, both FGF-19 and FGF-21 were significantly increased after bariatric surgery. FGF-19 did not correlate with insulin nor significantly with glucose and GLP-1 (supplemental Fig. 2). In this study, we also studied FGF-21 as a possible influencing factor in PBH. FGF-21 is known for improving insulin sensitivity and lowering blood glucose in mainly animal models [28]. FGF-21 did not differ between PBH and non-PBH patients.
To investigate a possible metabolic signature in the PBH group, we performed untargeted metabolomics. Several interesting metabolites surfaced in the analysis. We observed increased carnitine and in particular acylcholine levels in patients with PBH. Carnitine is a hydrophilic quaternary amine and is essential for beta-oxidation by transporting long-chain fatty acids over the inner mitochondrial membrane [29]. Carnitine accumulation may point to inhibition of carnitine palmitoyltransferase-1 (CPT-1) in the mitochondrial membrane. It is well known that insulin stimulates acetyl-CoA carboxylase leading to an increase in malonyl-CoA, the bonafide inhibitor of CPT-1. A decreased beta-oxidation will induce accumulation of long-chain fatty acyl-CoA. To salvage CoA, the cell may choose to transfer a choline moiety to the long-fatty acid, thus producing acylcholines. The metabolomics output contained linoleoylcholine, oleoylcholine, palmitoylcholine, arachidonoylcholine, eicosapentaenoylcholine, stearoylcholine, dihomo-linolenoyl-choline, and docosahexaenoylcholine. All were increased in the PBH group, suggesting that indeed this mechanism may be operative. Interestingly, next to acylcholines, plasmalogens were also increased. Acyl-CoA’s play a pivotal role in synthesis of plasmalogens [30]. We speculate that the increased levels of plasmalogens are induced by enhanced fatty acyl-CoA.
Could such a metabolic diversion explain the induction of PBH. The only clear difference between PBH and non-PBH was the increased insulin excursion. In combination with the restored insulin sensitivity after RYGB surgery, the increased insulin in PBH may just have tipped the balance and induced extra inhibition of beta oxidation, and as consequence increased glycolysis leading to decreased plasma glucose. Note that acylcholines have been suggested to interfere with acetylcholine signaling. It has been shown that pancreatic beta cells express the M3-muscarinic acetylcholine receptor. Mice in which the receptor was disrupted in the beta cell only show altered insulin signaling [31]. Acylcholines may interfere with this effect. Clearly these hypotheses require further investigation.
It is interesting to compare the metabolomic pattern in PBH with that observed for T2DM. In patients with diabetes, or insulin resistance, it is well known that fatty acyl-carnitines accumulate, indicating that mitochondria cannot cope with high influx of fatty acids in obese patients. This sequence of events is exactly opposite to what occurs in the PBH patients. Hence, the patients with T2D should show a decreased level of acylcholines and this was indeed observed [32, 33].
There are several limitations to our study such as the use of only two time points (0 and 120 min) for FGF-19 and FGF-21. Measuring the entire curve might have provided more insight in the interaction of these hormones with glucose. Second, we did not have repeated biopsies to investigate possible increased L-cell density after surgery [13, 15]. Finally, metabolomics data provided insight in relative concentrations, not absolute values of metabolites.
Collectively, our data showed that PBH might be caused by an impaired response to compensate for hypoglycemia by increasing gluconeogenesis. Carnitine accumulation, together with increased acylcholines, suggests impaired beta-oxidation possibly stimulating glycolysis in the PBH group. This novel insight may suggest new opportunities for therapeutic interventions against PHB.









留言 (0)