
記住我



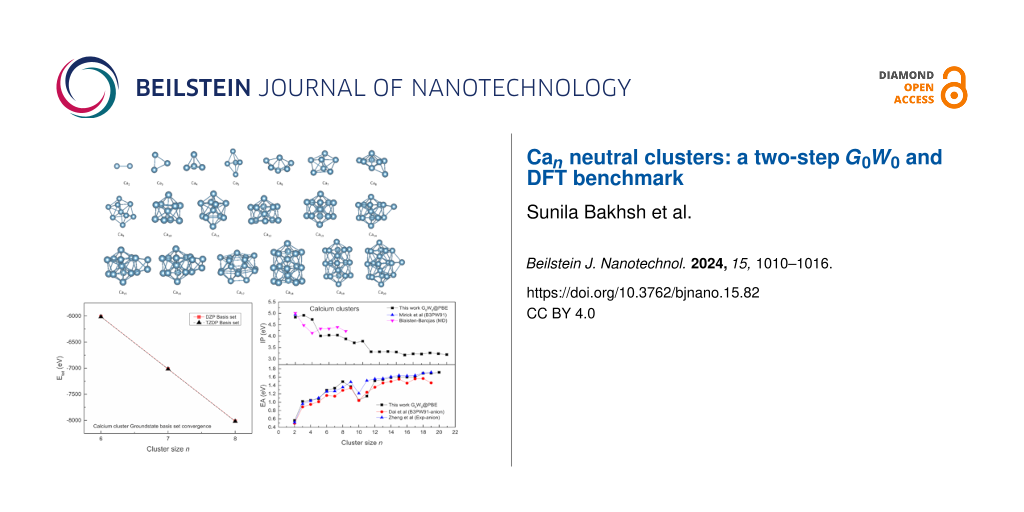

Calcium metal has a closed shell structure and belongs to the group IIA alkaline-earth metals . It has been widely used in carbon-chemical engineering (coating fullerene in hydrogen storage), optics, and materials science (as an ionic deposition) . The clusters of calcium are essential because they bridge the atomic and bulk materials; therefore, revealing their transition from micro- to macroscopic characteristics is a significant undertaking . Being a divalent metal, the size transition of calcium clusters occurs above the trimer cluster. Small calcium clusters (2–5) have been investigated using first-principles calculations incorporating the electron correlation effects . In 3D cluster structures, it can be used in the manufacturing of coating materials, which is useful for high-capacity hydrogen storage . As cluster size grows, the electronic configuration changes its semiconducting behavior from nonmetal to metallic due to the overlap between the s and p orbitals. Moreover, the geometry of clusters is related to their structural properties. Most of the studies on calcium clusters are limited to the dimer, where the binding energies and/or the ionization potentials (IPs) were determined spectroscopically . In addition to the experiments, a few theoretical DFT studies on calcium clusters focus only on metallic behavior, vibrational frequency analysis, and thermodynamic properties . However, there are no systematic studies on the electronic and structural properties of the series of calcium clusters.
Apart from some theoretical studies on calcium clusters, there are no systematic discussions on neutral cluster structural and electronic properties. Moreover, some studies have shown controversy in the binding energies of ground state clusters even for similar geometry and functionals used for calculation . In addition, there are no reported G0W0 studies for Ca clusters, which in the past have provided better IP and Egap for various systems. For small Ca clusters of up to 20 atoms, the structure, energies, and electronic structure were studied within the all-electron DFT approach. Our work aims to present the intricate characteristics of small Ca clusters by employing the DFT and state-of-the-art G0W0 approximation, which was recently used to predict the new ground-state structure of Be and Mg and successfully applied to obtain the correct IPs for these elemental clusters . This comprehensive benchmark study will help to enhance our understanding of these fascinating nanostructures and lead to their real-world utilization in various technological advancements . We used the DFT + GW scheme to investigate the electronic properties and structures of neutral Can (n = 2–20) clusters. From the DFT , one can obtain the accurate binding energies of the clusters, whereas the study of electronic properties from the GW approximation promises high accuracy in studying the excitation properties of the systems under consideration . Despite the wide availability of theoretical and experimental work on Ca clusters, no GW studies have been performed on neutral calcium clusters to the best of our knowledge.
Computational MethodHere, we employed particle swarm optimization (PSO) with CALYPSO code interfaced with ABACUS code to predict the neutral cluster of calcium (2–20) and local geometry optimization, respectively. The acquired structures were analyzed to determine among the low-energy isomers after running the calculation for 22 generations for calcium clusters (≈600 structures). Among the obtained 600 structures, the lowest four distinct isomers were selected for local geometry optimization for which GGA_PBE functional was used. The PBE functional is generally computationally less demanding compared to other functionals, as it can be more efficient in terms of computational resources and time. The threshold for the force was set at 0.1 eV/Angstrom for better convergence, whereas the charge density difference tolerance, which is essential for convergence, was set at a value of 10−9. The ABACUS code employs the ONCV-type multi-projector pseudopotentials for the description of the core ions, which is used in our calculation as provided by the SG15 library . We have therefore set the energy cutoff as 100 Ry for better accuracy. The double-ζ plus polarization (DZP) basis set was used in ABACUS calculations, which was tested against the triple-ζ plus double polarization (TZDP) to obtain the total energies of Ca6, Ca7, and Ca8. The convergence plot can be found in Supporting Information File 1, Figure S1. It is worth mentioning that our motivation for the selection of DZP is based on our previously reported data for DZP and TZDP basis sets, which were tested for various isomers of Be and Mg clusters. These results have shown that the basis sets are well converged and even DZP is sufficient to obtain reliable results .
To study the energetics of Can clusters, we used the G0W0 calculations in the FHI-aims code . For the G0W0 calculations in the FHI-aims code, the NAO basis sets are employed with PBE functional to relax the structural geometry using the tier 4 and “tight” settings, providing a better description of IPs and HOMO–LUMO gaps of the molecules and clusters. In addition, the G0W0 calculations are performed to obtain GW-calculated eigenvalues using the two-pole fitting by setting 40 frequency points.
Results and Discussion Structural geometryThe geometries of the ground-state structures of Ca clusters (2–20) were analyzed by the ABACUS software and are displayed in Figure 1.
![[2190-4286-15-82-1]](https://www.beilstein-journals.org/bjnano/content/figures/2190-4286-15-82-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The ground-state structures of Ca clusters (n = 2–20).
The geometrical structures predicted in this work, with a few exceptions similar to those obtained by the work of Liang et al. . The trimer forms an equilateral triangle structure, whereas the three-dimensional configurations are more favorable for larger clusters. Figure 2 shows the binding energies of the cluster for the size range of 2–20 in comparison with reported data which is calculated from Equation 1:
![[2190-4286-15-82-i1]](https://www.beilstein-journals.org/bjnano/content/inline/2190-4286-15-82-i1.svg?max-width=590&scale=1.18182) (1)
(1)Here, Etot is the total energy of the cluster after the relaxation step, n is the cluster size, and Eatom represents the free atom energy.
![[2190-4286-15-82-2]](https://www.beilstein-journals.org/bjnano/content/figures/2190-4286-15-82-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The binding energies of ground-state Ca clusters (n = 2–20) in comparison with reported data .
The experimental studies of calcium dimers showed that the ground state of Ca2 has a low binding energy of 0.14 eV. Our dimer results are also significantly closer to the reported experimental and theoretical work . The binding energies also increase rapidly up to cluster size n = 7. A decrease in binding energy is seen at n = 8, likely due to the shell closing effect. Ca7 might represent such a stable configuration that an extra atom has to form Ca8 which could mean starting a new electronic shell and is less stable initially, resulting in a drop in binding energy. In addition, the structural geometry can play a crucial role in the stability of a cluster. Ca8 is a pentagonal bipyramid structure with a capped atom, which disrupts its symmetry from a perfect pentagonal bipyramid. This symmetry breaking can also be a possible reason, as it might generate a structure with increased surface strain or less favorable bonding environments. Apart from this, we can also analyze Ca7 whose binding energy is much higher in the series of clusters as shown in Figure 2. The higher binding energy depicts higher stability, which can be confirmed by the second difference analysis (see Figure 3). Another possible reason for the drop in binding energy from n = 7–8 can be linked to the shift from a more stable structure of Ca7 to a less stable structure of Ca8 (as the symmetry is disrupted by adding a capping atom). This observation was not seen in previous studies and could be due to the difference in the type of functional and code used in the calculation. The use of hybrid functionals generally improves accuracy for many systems, but it does not guarantee perfect results for all types of bonding or electron correlation effects. Overall, the accuracy of theoretically obtained structures is supported by the experimental data, which is lacking in this case. Apart from the accuracy of the functional, there can be a van der Waals interaction effect for clusters, which can be calculated by semi-empirical corrections added to the conventional density functional approximation and needs detailed assessment for small clusters (n = 2–10). In such cases, DFT-D methods can be used along with the standard DFT calculations.
![[2190-4286-15-82-3]](https://www.beilstein-journals.org/bjnano/content/figures/2190-4286-15-82-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The second difference in energy for calcium ground-state clusters (n = 2–20) The data is plotted against the reported work of Liang et al. and Dai and Blaisten-Barojas .
By further increasing the cluster size, the binding energy smoothly increases again. The PBE results are plotted in comparison with the reported experimental work of Balfour et al. and theoretical works . Comparing the reported theoretical work, we can see some inconsistencies in the predicted binding energies at cluster size n = 8–11. In our work, the reported binding energies depict a sharp decrease in Eb, which is missing in other theoretical literature. In our case, the cluster size n = 8, which is a pentagonal bipyramid structure with an added atom to break its symmetry, can be a reason for a sudden drop in the cluster binding energy; however, a decrease or any change can also be attributed to the dispersion effect. Overall, the binding energies reported here are higher compared with all theoretically reported data. This difference can be due to two reasons. First is the use of a different functional and second are the basis sets and accuracy, which are different for the reported data compared with our work. In addition, van der Waals dispersion effects can also affect the binding energies of clusters, which need semi-empirical corrections . We have also drawn a polynomial fit to calculate the convergent value of binding energy; however, the resulting value is 1.35 eV for binding energy per atom in Ca. This obtained value is small, as compared to the bulk value, which is 1.825 eV. In addition to the binding energies, the second difference in the energy analysis was carried out to analyze the stability of the geometrical structures, as shown in Figure 3.
It can be seen from the above figure that 2, 5, 7, 9, 10, 11, 14, 17, and 19 have positive values for the second energy difference. This indicates that these clusters are relatively stable among their neighboring clusters. In addition, it is seen that the clusters with sizes n = 4, 8, 12, 15, and 18 have negative second differences in energies, suggesting that these clusters are less stable among the series (see Figure 3). Moreover, clusters such as Ca7 and Ca9 have higher stability. The higher stability of these two clusters can be described by the ellipsoidal shell distortions that can occur in metal clusters . A similar case was seen in our previous study of magnesium clusters, where the clusters with sizes n = 7, 15, and 17 showed relatively high stability from the second difference analysis . In such cases, the predicted equilibrium shape of the cluster is ellipsoidal instead of spherical. It is also evident that the magic cluster Ca10 can be seen to have a lower positive second difference compared to other clusters in the series. The cluster exhibits a slightly distorted C3v structure. In the case of Ca10, its deviation from a perfect tetrahedral structure could be a contributing factor to its relatively modest second difference in energy.
Electronic propertiesThe electronic properties of Ca clusters have been studied by the all-electron code FHI-aims, and the simulations are performed at the G0W0@PBE level. The IPs and EAs are presented in Figure 4 along with the reported data. Our dimer ionization potential, which is 4.83 eV for Ca clusters from G0W0@PBE is closer to the theoretical value of 4.95 eV . In the upper panel of this figure, there is a decreasing pattern for the calcium cluster for ionization potentials. For the magic cluster Ca10, a larger ionization potential is obtained compared to the other neighboring clusters of the series. A swift fall in the ionization potential occurs for cluster Ca11, which shows the shell closing at n = 10. For larger clusters, the ionization potential curve becomes relatively smooth. Normally, as the cluster size increases, the IP tends to decrease. The smaller clusters tend to have higher IPs because electrons are more tightly bound. On the other hand, the larger clusters have more delocalized electrons, which results in lower IPs in these clusters. Clusters with lower IPs are generally more reactive, as they can more easily lose an electron. In our work, we have observed peaks at ≈4.83 and 4.90 eV. Such peaks suggest that these clusters are more stable than their neighboring clusters. The comparison with reported anionic clusters is not possible due to the unavailability of any data for EAs for neutral clusters. A sharp decrease in the EAs is obtained for Ca10, whereas the ionization potential for this cluster is relatively high, which indicates it is a highly stable magic cluster. Our G0W0 IPs and EAs for the neutral cluster are reasonably close to the available data. Moreover, the EA shows an increasing trend, which means larger clusters have more electron affinity than smaller ones. For IPs, the Jellium even and odd oscillations except for a few clusters are not prominent for calcium clusters. This may suggest that the electron interactions in such clusters are more localized compared to the delocalized electron sea assumed in the Jellium model.
![[2190-4286-15-82-4]](https://www.beilstein-journals.org/bjnano/content/figures/2190-4286-15-82-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The IPs and EAs of calcium ground-state clusters (n = 2–20). The data is presented by G0W0 studies, compared with anionic experimental work and theoretical reports .
Another remarkable aspect of the cluster studies is to understand whether there is an underlying correlation between the ionization potential and electron affinities of the clusters. To study the correlation between the IPs and EA (as the function of cluster size), we applied a linear regression to analyze the quantitative estimation of the strength of the correlation for Ca clusters. For the binding energies versus the IPs, linear regression analysis returned the R2 value of 0.85. On the other hand, for binding energies versus EAs, we obtained the R2 value of 0.76. These results suggest there is a stronger correlation of binding energies Eb with IPs than EA. Moreover, the linear regression analysis for Eb versus Egap yielded the R2 value of 0.89, which is the highest of all three. This suggests that the strongest correlation exists between the binding energies and Egap for Ca clusters. In addition, we also plotted the second differences of the binding energies with the IP, EA, and Egap which can be seen in Supporting Information File 1, Figure S2a–c. All three results in this figure show that the second difference between IPs and EAs closely follows Eb indicating a good correlation. Supporting Information File 1, Figure S2c indicates the relationship between the relative thermodynamic stability of the clusters and Egap which is also called “the maximum hardness principle” . From these three results we can see that the oscillatory pattern is moderated with the evolution of the cluster size. However, the majority of the values of IPs, EAs, and Egap are positive and following closely which suggests a clear correlation.
ConclusionWe presented a DFT and G0W0 combined approach to study the electronic and structural properties of neutral calcium clusters for a size range of n = 2–20. The initial geometries are obtained with the CALYPSO structure prediction algorithm, interfaced with the ABACUS code. The electronic properties, specifically the IPs and the EAs of Ca clusters, are calculated with the all-electron code FHI-aims with the G0W0 scheme. From the structural stability study, the second-difference analysis of the consecutive clusters for both binding energies demonstrated the structural stability of clusters such as Ca2, Ca5, Ca7, Ca9, Ca11, Ca14, Ca17, and Ca19, in addition to Ca10, which is also a magic cluster. The ionization potentials for Ca clusters from the G0W0 scheme are reported for the first time. Moreover, the high ionization potentials are obtained by the state-of-the-art G0W0 approach at a particular cluster size; in this case, Ca2, Ca3, Ca10, Ca14, and Ca18 clusters, which suggests that to ionize these clusters, more energy is required. Hence, one can suggest that energetically, the most stable structures do not necessarily have the largest IPs or EAs. Additionally, clusters with a larger Egap should have a higher abundance, which suggests they should be chemically more stable. In summary, predicting the ionization potentials adds to our understanding of the electronic structure and energetics of the calcium clusters. This benchmark may provide useful insights for future exploration of size-dependent properties and their potential applications, such as developing theoretical models and designing functional materials.
留言 (0)