
記住我




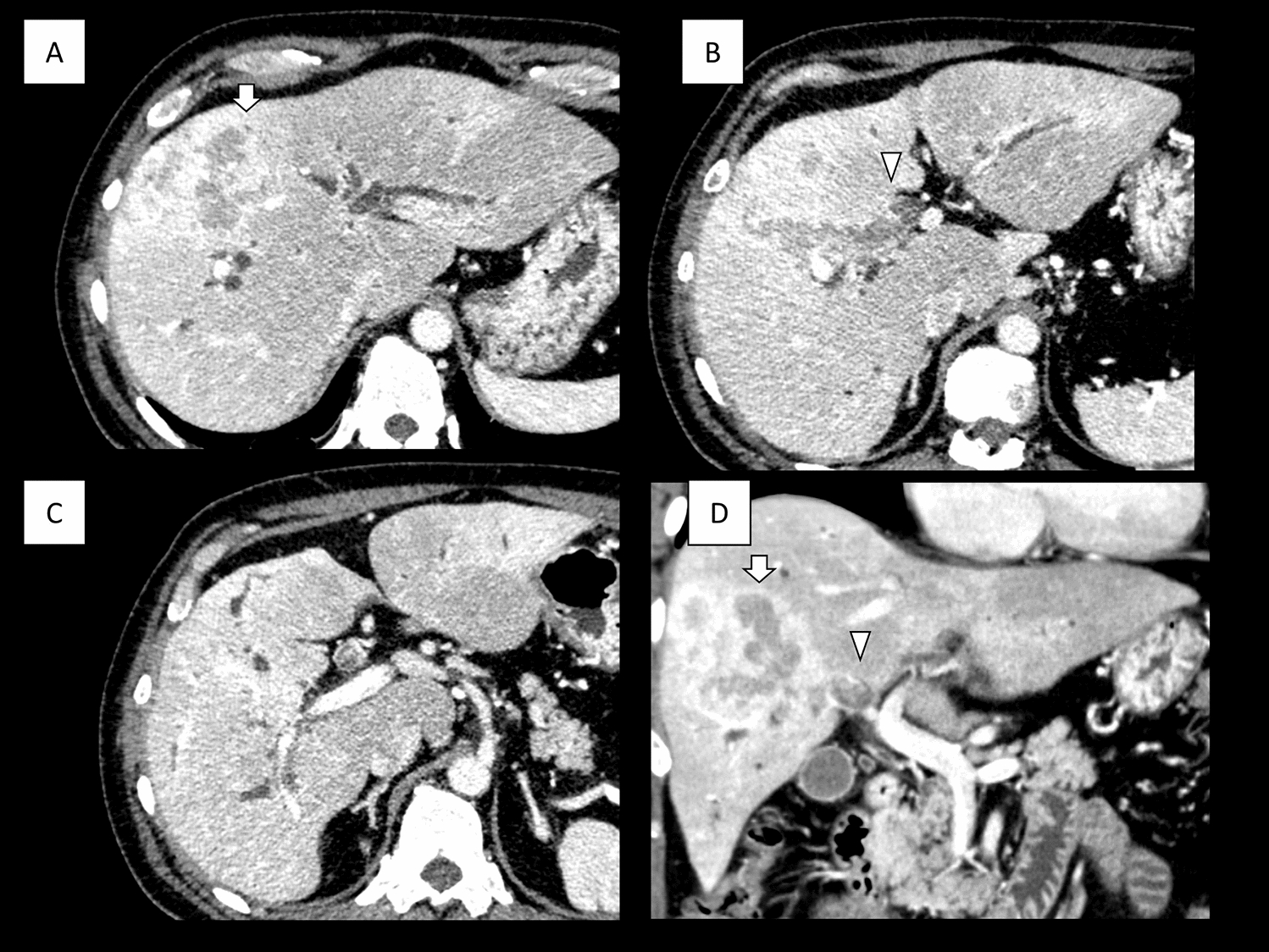




To distinguish LS from the less common familial adenomatous polyposis, the term hereditary nonpolyposis colon cancer (HNPCC) was coined, and for many years the terms Lynch syndrome and HNPCC were used interchangeably. Earlier diagnosis of LS with high penetrance may have explained the history of early-onset CRC in the presented patient. To capture more families with hereditary cancer, the Amsterdam II Criteria was developed. These were based on the same criteria as Amsterdam I but were expanded to include several tumors associated with HNPCC in addition to CRC. Currently, the term HNPCC has been discouraged, because these patients do, in fact, make polyps, although not as many as is typical in Lynch syndrome [6]. Lynch syndrome must be distinguished from other familial adenomatous polyposis syndromes. The term familial colorectal cancer type X refers to the nearly 40% of patients who satisfy the criteria for Amsterdam I but do not have a genetic profile consistent with Lynch syndrome [6]. To date, however, a specific coding defect has not been identified to differentiate familial CRC type X at-risk family members by formal genetic testing. Nevertheless, further genetic testing in the presented patient revealed not only one mutated copy of her MSH6 MMR gene but an additional mutation on the opposing allele, establishing the diagnosis of CMMR-D. The most common initial approach uses immuno- histochemistry or microsatellite instability testing. Based on the results, patients can then be appropriately triaged and tested for formal germline defects. Mutated copies need to be passed on through meiosis, therefore each parent consequently must have been a carrier (Fig. 4).
Fig. 4
Genetic transmission of MMR mutations. To acquire HNPCC, recipient must receive one mutated copy of AD MMR mutation. CMMR-D offspring require two copies of mutated gene as they are inherited in AR fashion. The patient at discussion has parents that must both contain mutations in repair genes to have an established diagnosis of CMMR-D. [3]
CMMR-D is a rare syndrome with an incidence of 1/million patients [3]. Its infrequent occurrence has created a lack of awareness and often goes unnoticed until cancer occurrence and progression are diagnosed in young patients. Frequently associated neoplasms include colon (70%), brain (70%), small bowel (10%), and lymphoma/ leukemia (20–40%). In addition, patients with biallelic mutations in MSH6 have a significantly higher incidence of brain tumors manifested mainly in the first 10 years of life when compared to other mutations [6, 7]. MUTYH-associated polyposis syndrome (MAP) can be characterized as an autosomal recessive germline mutation in the MUTYH base excision repair gene and leads to an increased risk for gastrointestinal cancers [5]. Multiple adenomas can be found in the digestive tube from accumulated mutations leading to CRC. Although, the patient has not been diagnosed with MAP, this specific MUTYH mutation was confirmed in her genetic testing, placing her at risk for CRC. The former mutation confers a 28-fold increase in their likelihood of developing CRC [5, 8]. Currently, there are no absolute treatment options for those with CMMR-D. Prevention by continuous surveillance for early disease detection remains the best measure to implement proper treatment for an increase in lifespan with improved quality of life.
CRC presenting early in life should raise clinical suspicion of CMMR-D, especially those associated with brain, hematologic, and endodermal origin. Detailed exploration of family history and sibling presentation can be beneficial. Assessment in the history of pediatric/young adult malignancies may provide a score guiding indications for early surveillance and genetic testing in those with characteristics of CMMR-D. Once the diagnosis of CMMR-D is established, we recommend a multidisciplinary oncologic team to follow the patient with emphasis in the annual screenings of the GI, endometrium, and urinary tract. Blood panel and brain MRI should be done every 6 months.
留言 (0)