
記住我




Inflammatory bowel disease (IBD) is a destructive, long-lasting, and immune-mediated disease, mainly including crohn’s disease (CD) and ulcerative colitis (UC) (Jiang et al., 2022). Despite significant advances have been made in exploring the occurrence and development of IBD, the exact pathogenesis is yet unclear. Immune dysfunction, intestinal dysbiosis, genetic susceptibility alongside environmental triggers may contribute to the development of IBD (Abraham and Cho, 2009; Dang et al., 2023) (Figure 1). It’s universally acknowledged that IBD is a global disease with high incidence and prevalence (Kaplan and Windsor, 2021). The chronic inflammation and remission-relapse pattern of IBD make patients experience chronic abdominal pain and repeated diarrhea, which exerts a significant impact on the quality of life (Chen et al., 2024). Available data indicated that the cumulative rates of hospitalization in CD and UC patients were 23%–49% and 9%–33% at 1 year; the 5-year hospitalization rates ranged between 44% to 54% and 18% to 54% for CD and UC, respectively. During the first 5 years after diagnosis, the cumulative rates for surgery were 5%–10% for UC and 10%–40% for CD. What should be noted is that the risk of developing colorectal cancer in patients with UC was two times higher than general population (Zhao et al., 2021). The high hospitalization and surgery rates, as well as high risk of developing cancers significantly increase medical costs for patients with IBD. In 2017, the global disability-adjusted life-years caused attributed to IBD was 1.85 million, about 1.5 times as that in 1990 (1.25 million) (GBD, 2017 Inflammatory Bowel Disease Collaborators, 2020). Indeed, it poses a huge burden on global healthcare systems.
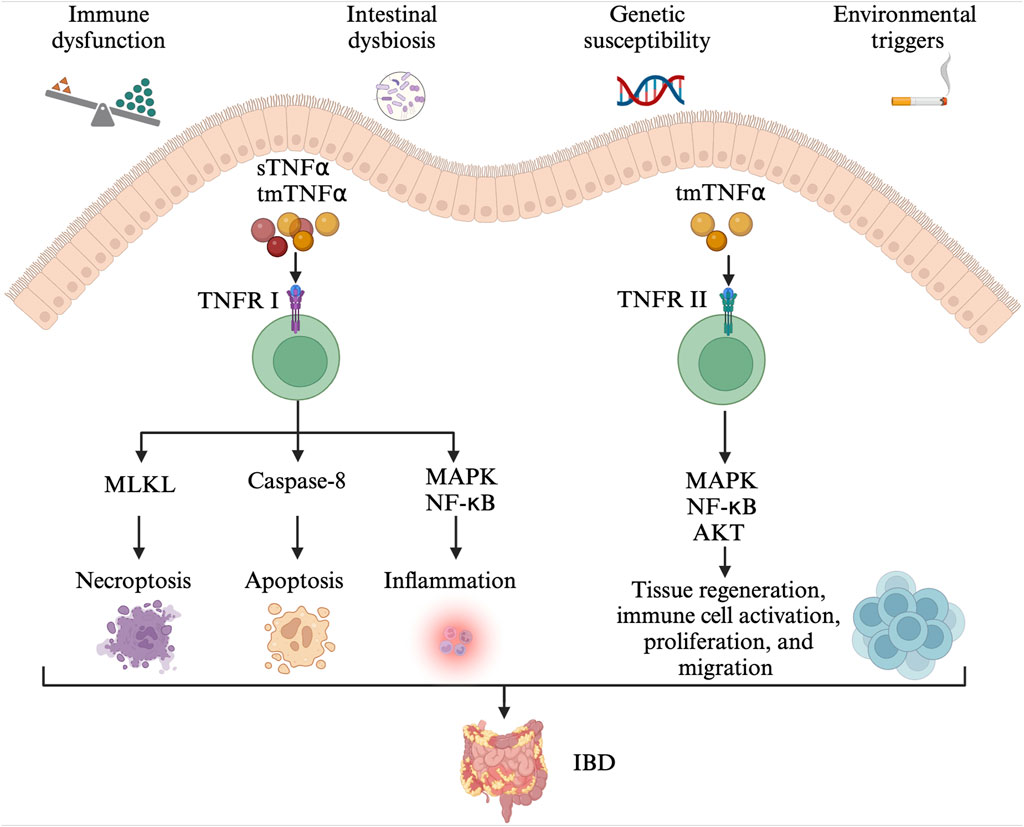
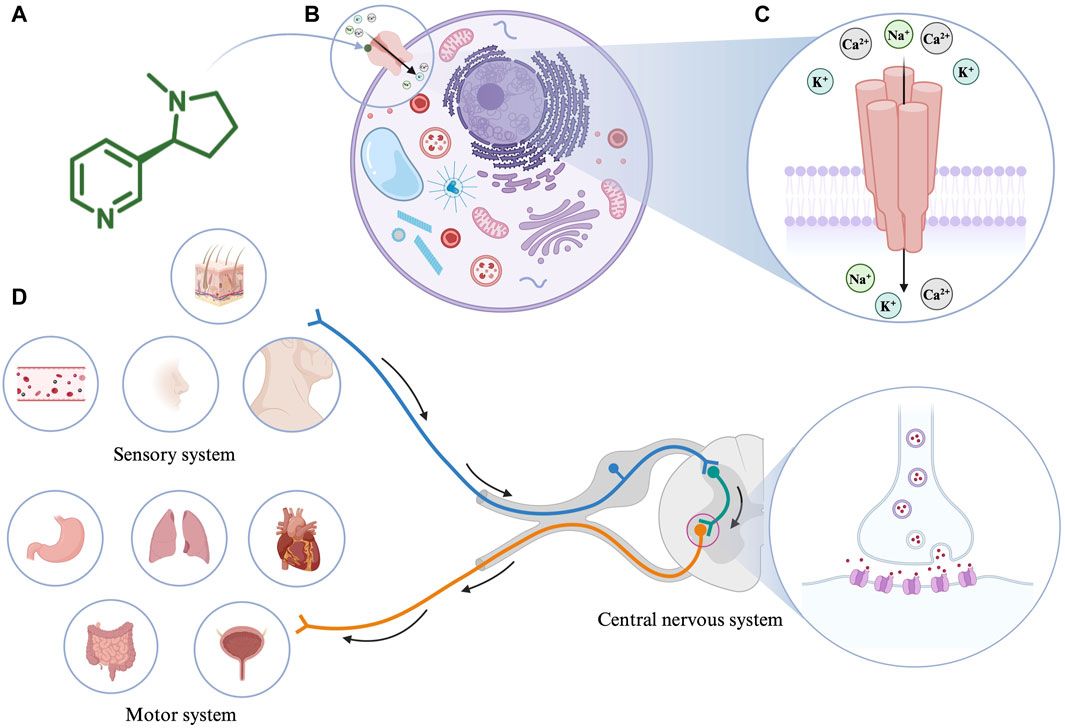
Figure 1. The pathogenesis of IBD. Immune dysfunction, intestinal dysbiosis, genetic susceptibility alongside environmental triggers contribute to the development of IBD. The tmTNFα and sTNFα bind with TNFRI, mediating the activation of pro-inflammatory (MAPK and NF-κB) signaling pathways, MLKL-dependent necroptosis, and caspase-8-dependent apoptosis. The TNFR2 signaling pathway is mainly activated by the tmTNF-α. The tmTNFα binds with TNFRII and activates MAPK, NF-κB, and AKT signaling pathways, involved in tissue regeneration, immune cell activation, migration, and proliferation. IBD: inflammatory bowel disease; tmTNFα: transmembrane TNFα; sTNFα: soluble TNFα; TNFRI: TNF receptor I; MAPK: mitogen-activated protein kinase; NF- NF-κB: nuclear factor kappa-B; MLKL: mixed-lineage kinase domain-like protein; TNFRII: TNF receptor II.
Available data indicated that the mean healthcare costs for CD and UC were $8,265 and $5,066 per patient-year in the United States in 2004, respectively (Kappelman et al., 2008). From 2007 to 2016, the direct healthcare costs for IBD (CD and UC) increased to $22,987, three times higher than non-IBD controls ($6,956) (Park et al., 2020). In Europe, the mean total healthcare costs for CD and UC rose from €2,548 and €1,524 per patient-year in 2003 to €3,500 and €2,000 in 2020, respectively (Odes et al., 2006; Zhao et al., 2021). In China, the mean direct care costs for IBD (CD and UC) are $7,944 per patient-year from 2018 to 2019 (Yu et al., 2021). In the initial stages, the major drivers of healthcare costs for IBD were hospital and surgery. However, with the rapid progress made in drug development, the main health costs have shifted to medication. The global IBD medication treatment market size is extremely large. The introduction of biologics innovated IBD treatment and thus accounted for the majority of healthcare expenditures. Available data showed that biologics accounted for €1,782 for CD and €286 for UC per patient-year in Europe (Burisch et al., 2020). Anti-tumor necrosis factor-α (anti-TNFα) is the first approved biologic agent for CD and UC (Buchner et al., 2021). Among these biologics available for IBD, the annual costs of anti-TNFα treatment are considerable, making up 64% and 31% of the total costs in CD and UC, respectively (van der Valk et al., 2014). Although some anti-TNFα biologics have been included in medical insurance, the financial burden of IBD, especially for anti-TNFα biologic drugs, is still heavy. The high price further limits the access to anti-TNFα biologic treatment in resource-limited settings.
TNFα is a pro-inflammatory cytokine and plays an important role in the pathophysiology of IBD (Figure 1) (Chen L. et al., 2020; Chen et al., 2021). TNFα exists in two forms, the transmembrane and soluble form. On the one hand, transmembrane TNFα (tmTNFα) and the soluble TNFα (sTNFα) can bind with TNF receptor I (TNFRI), mediating the activation of mitogen-activated protein kinase (MAPK) and nuclear factor kappa-B (NF-κB) signaling pathways, and then, producing pro-inflammatory cytokines, cell adhesion molecules and synthetase nitric oxide (Wang and Shen, 2022). The binding between them also can activate caspase-8-dependent and mixed-lineage kinase domain-like protein (MLKL) death signaling pathways, involved in apoptosis and necroptosis, respectively (Jang et al., 2021). On the other hand, the binding of tmTNFα with TNF receptor II (TNFRII) can also activate MAPK, NF-κB, and AKT signaling pathways, causing tissue regeneration, immune cell activation, migration, and proliferation (Levin et al., 2016; Zeng et al., 2023). As a result, severe intestinal inflammation and mucosal barrier injury occur. In order to prevent its pro-inflammatory process, monoclonal antibodies to TNFα including infliximab, adalimumab, golimumab, and certolizumab have been developed and approved for CD and/or UC treatment (Leone et al., 2023). They may exert their therapeutic effects in the induction and maintenance of disease remission by inducing CD4+ T cell apoptosis and/or promoting the differentiation from monocytes to M2-type wound-healing macrophages (Levin et al., 2016).
Despite anti-TNFα biologics show favorable therapeutic effects in achieving clinical, endoscopic, and histologic remission in IBD, the annual costs are really high (Jiang et al., 2023). The expiration of patents of some anti-TNFα biologics has further facilitated the development of biosimilar agents. Biosimilars potentially reduce the high costs of biologics and increase patient access to biologics due to the stiff competition in the pharmaceutical market and extrapolation across indications (Fiorino and Danese, 2014). In this review, we briefly introduce the drug utilization, effectiveness, and safety of the most used anti-TNFα originators (infliximab and adalimumab), and elaborate on the efficacy and safety of these biosimilars in IBD. Furthermore, we also evaluate the efficacy and safety of the switches from originators to biosimilars, and discuss the benefits, challenges, and future directions of biosimilars in IBD.
2 The use of anti-TNFα originators in IBD2.1 What are anti-TNFα originatorsAnti-TNFα originators, discussed in this review, are the two anti-TNFα biologicals. Although biologicals comprise various groups of medicines, such as monoclonal antibodies, vaccines, growth factors, immune modulators, and medicines derived from human blood. Our review mainly discusses the two anti-TNFα monoclonal antibodies (infliximab and adalimumab). Anti-TNFα monoclonal antibodies are purified from human or mouse living systems, completely different from small molecules that are produced by chemical synthesis or purified from plants (Buchner et al., 2021). Anti-TNFα originators are a diverse group of original, independent research and development new drugs with pharmaceutical patents, usually used as licensed reference products (Kang et al., 2023). Anti-TNFα originators follow a complex and long process for regulatory approval, including drug screening and optimization (structure, pharmacologic action, and biological activity), preclinical studies (pharmacokinetics, pharmacodynamics, and toxicology in vitro and in vivo studies), clinical studies (I–III randomized clinical trials), marketing approval, and post-marketing research (IV clinical trial), which significantly increases the time and money costs. Besides, the manufacturing costs of anti-TNFα originators are very high. Available data indicated that the cost to develop a new biological agent is about $2.0 billion, significantly higher than the production costs of biosimilars ($100–250 million) (Zheng et al., 2017).
2.2 InfliximabInfliximab, a human-mouse chimeric anti-TNFα monoclonal IgG1 antibody, is the first biologic approved for CD by the United States Food and Drug Administration (FDA) in 1998. It binds with TNFα and prevents the binding between TNFα and TNFR (Knight et al., 1993). Until now, it has been approved for various indications including CD, UC, rheumatoid arthritis (RA), psoriasis, and others. The famous ACCENT I randomized trial of 573 moderate to severe CD patients showed that infliximab can induce disease response at week 2 in 58% (335/573) of patients. At week 30, the clinical remission rates were higher in the infliximab maintenance group (5 mg/kg infliximab and 10 mg/kg infliximab), compared with the placebo group (39% vs. 21%, 45% vs. 21%, respectively). The maintenance treatment efficacy of infliximab was also claimed at week 54. At week 54, the proportion of patients who discontinued corticosteroid treatment in the infliximab maintenance group was 2.22 times higher than that of the placebo group (29% vs. 9%). Besides, patients in the infliximab maintenance group also presented lower mean Crohn’s Disease Activity Index (CDAI) and higher mean inflammatory bowel disease questionnaire (IBDQ) scores (Hanauer et al., 2002). Recently, a network meta-analysis of 25 clinical trials and 8,720 CD patients claimed that infliximab had optimal efficacy in the induction of clinical remission in patients with luminal CD (Barberio et al., 2023). The excellent therapeutic effects of infliximab were also confirmed in another ACCENT II trial of 306 fistulizing CD patients. In comparison with the placebo group (19%), 36% of patients with infliximab treatment had a total absence of fistulas at week 54 (Sands et al., 2004). As for the safety of infliximab, the FDA label indicated that the risk of serious adverse events (SAEs) including serious infections and malignancy increased in the infliximab treatment group, although SAE rates were similar between the infliximab treatment arm and the placebo arm in another study (Sands et al., 2004; Food and Drug Administration, 2021). In a word, infliximab treatment is effective and safe in inducing and maintaining disease remission in moderate to severe CD.
In UC, the Active Ulcerative Colitis Trials 1 and 2 (ACT 1 and ACT 2) of 364 moderate to severe UC patients revealed that infliximab therapy can induce clinical remission and mucosal healing as early as week 8, and maintain effective during week 54 (Rutgeerts et al., 2005). As for patients with steroid-refractory acute severe ulcerative colitis (ASUC), infliximab outperformed cyclosporine in achieving endoscopic remission at day 98 (73% vs. 25%) (Laharie et al., 2021). Moreover, infliximab was also claimed to be an effective salvage treatment for patients with tacrolimus-refractory ASUC (Yamamoto et al., 2010). It was also proven to be safe in treating UC with regard to similar rates of adverse events (AEs), infections, and acute infusion reactions (Rutgeerts et al., 2005). It should be noted that infliximab is a chimeric antibody, implying a higher possibility of formation of antibodies to infliximab. As a result, the risk of experiencing infusion reactions and even loss of efficacy may increase (Su and Lichtenstein, 2003). Concomitant immunosuppressive therapy or changing antibody structure may mitigate immunogenic responses (Su and Lichtenstein, 2003). Indeed, the introduction of infliximab innovated IBD therapy and became the mainstay of treatment for refractory IBD. Further studies on other anti-TNFα biologics are therefore encouraged.
2.3 AdalimumabAdalimumab is also an anti-TNFα monoclonal IgG1 antibody, but it is different from infliximab regarding antibody structure (Tracey et al., 2008). It is a fully human, recombinant monoclonal antibody with lower immunogenicity and a larger antigen-antibody interface (Tracey et al., 2008; Hu et al., 2013; Kennedy et al., 2019). The CLASSIC-I trial of 299 moderate to severe CD patients (naive to anti-TNFα antagonists) claimed that the adalimumab 160/80 treatment (160 mg at week 0 and 80 mg at week 2) was more effective than placebo in inducing clinical remission (36% vs. 12%) (Hanauer et al., 2006). One year later, the CLASSIC II trial further demonstrated its significant efficacy and safety in maintaining clinical remission during week 56. In comparison with the placebo group, the adalimumab treatment group (40 mg every other week) presented higher remission rates (79% vs. 44%), greater mean decreases of CDAI scores (197.7 vs. 119.6), and higher IBDQ scores (Sandborn et al., 2007). In the same year, the better therapeutic effects of adalimumab were also found in those CD patients previously exposed to anti-TNFα therapy. This finding suggested that adalimumab could be an additional treatment option for those who lost response to and/or were intolerant to infliximab. Besides, patients receiving adalimumab were more likely to achieve corticosteroid-free remission and fistula remission than the placebo group (Colombel et al., 2007). Recently, the CREOLE study further evaluated the efficacy of adalimumab in CD patients with symptomatic small bowel stricture (SSBS) and proved its excellent effects in patients with SSBS due to CD. Treatment with adalimumab can make 53% of patients free of surgery 4 years after initiation (Bouhnik et al., 2018). A large meta-analysis of 31 clinical trials recommended adalimumab as second-line therapy for patients who were intolerant to infliximab (Singh et al., 2021).
As for moderate to severe UC patients, a multicenter study of 576 patients suggested that the clinical remission rates at week 8 in the adalimumab subcutaneous injection regimen arm (160 mg at week 0 and 80 mg at week 2) were one time higher than that in the placebo group (18.5% vs. 9.2%) (Reinisch et al., 2011). At week 52, 17.3% of patients in the adalimumab group maintained clinical remission, compared with 8.5% of patients in the placebo group. Moreover, more patients with adalimumab therapy achieved sustained mucosal healing (at week 8 and week 52), and sustained corticosteroid-free remission (at week 32 and 52) than the placebo group (18.5% vs. 10.6%, and 10.0% vs. 1.4%, respectively) (Sandborn et al., 2012). Even in those patients with a history of anti-TNFα therapy, the adalimumab treatment group was more likely to maintain sustained clinical response at week 8, and week 52 than the placebo group, providing an alternative therapeutic option to patients who experienced infliximab failure (Sandborn et al., 2012). A cost-effectiveness analysis from the United Kingdom further claimed that the total costs (including costs of drug acquisition and administration, direct and indirect healthcare costs) and biologic costs for adalimumab was lower than infliximab (£194,764.73 vs. £206,065.90, and £10,289.40 vs. £19,285.37, respectively). And patients on adalimumab incurred slightly higher quality-adjusted life years (QALYs) than those on infliximab treatment (13.872 vs. 13.788) (Wilson et al., 2018). Caution needs to be taken when interpreting these results, because different study designs, different standards of treatment response, and different ethnicities are used in various studies. Collectively, adalimumab is an effective and well-tolerated biologic drug for moderate to severe CD and UC patients. It also became the efficacy benchmark of its category and the reference product for bioequivalence studies.
3 The use of anti-TNFα biosimilars in IBD3.1 What are anti-TNFα biosimilarsBiosimilars are biological products that are similar in terms of quality, safety, and efficacy to an already licensed reference product (World Health Organization, 2022). Therefore, the anti-TNFα biosimilars are a group of monoclonal antibodies that contain a version of the active pharmaceutical ingredient and associated molecules of already licensed original biologics (originators) (Blandizzi et al., 2017; World Health Organization, 2022). Anti-TNFα biosimilars are different from generic medicines in terms of the drug substance. The former contains similar active ingredients, while the latter has identical active ingredients (Blandizzi et al., 2017). Moreover, given the relatively high molecular weight, complicated three-dimensional protein structure, and complex posttranslational modification, the structural sameness and bioequivalence evaluation approaches used in generic medicines is not applicable to biosimilars. Firstly, researchers should characterize the quality attributes of the reference product, and make direct head-to-head comparison between the licensed reference product and the biosimilar in terms of structural and functional similarity (in vivo and in vitro). Then, the clinical pharmacologic comparability assessment (pharmacokinetic modeling, pharmacodynamic modeling and immunogenicity) is carried out in one or more indications (if possible). Then, the comparative clinical trials (safety, efficacy, and immunogenicity profiles) are performed in one or more sensitive populations (if possible) (Lyman et al., 2018; World Health Organization, 2022) (Figure 2).
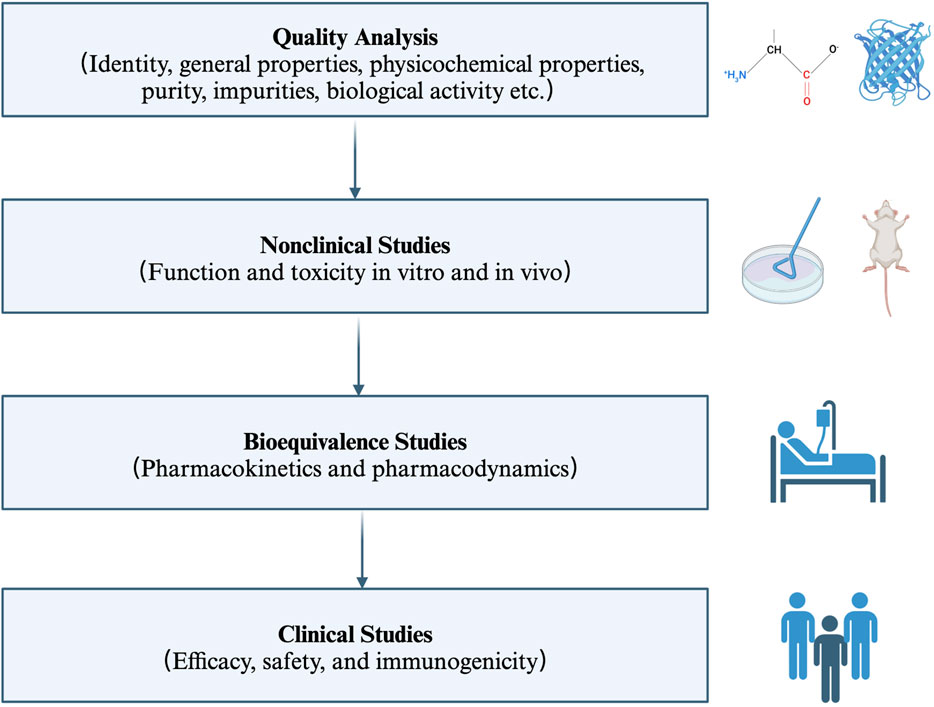
Figure 2. Key principles for the licensing of biosimilars by WHO. The regulatory and approval pathway for biosimilars includes four steps. Firstly, characterize the quality attributes of the biosimilar and the reference product. Secondly, evaluate the pharmaco-toxicological activity of the biosimilar and the reference product in vitro and in vivo; Thirdly, investigate the pharmacokinetic and pharmacodynamic profiles in healthy volunteers; Fourthly, assess the efficacy, safety, and immunogenicity profiles in one or more indications. WHO: World Health Organization.
The regulatory process for approval of anti-TNFα biosimilars is speedier and easier in comparison with originators. The core evidence to support regulatory approval for anti-TNFα biosimilars is obtained from manufacturing and preclinical data. While the marketing approval for originators depends more on extensive clinical data. Besides, extrapolation across indications further accelerates the regulatory approval process of anti-TNFα biosimilars. Once clinical bioequivalence is fulfilled in one condition, this biosimilar may be approved for other indications for which the reference product has been approved, without the need for repeating clinical trials across different indications. This process is called extrapolation (Lyman et al., 2018). Most clinical equivalence studies of anti-TNFα biosimilars have been conducted in patients with RA and/or those with plaque psoriasis, rarely in patients with IBD (Ben-Horin et al., 2016). Collectively, anti-TNFα biosimilars follow an accelerated process for marketing approval.
The biosimilar, Omnitrope, was approved by the European Medicines Agency (EMA) for patients with growth hormone deficiency in April 2006 (Fuhr et al., 2010). It is the first biosimilar approved for patients. Seven years later, biosimilars of infliximab, Remsima and Inflectra (CT-P13) got approval for CD or UC in September 2013. They two have become the first monoclonal antibody biosimilar approved by the EMA. In 2016, Inflectra was firstly approved by the FDA for patients with IBD. One year later, the biosimilar of adalimumab, Amjevita (ABP 501) was approved for patients with IBD. In recent 10 years, several other biosimilars of infliximab such as Flixabi (SB2) and Zessly (PF-06438179/GP1111) (Table 1), and biosimilars of adalimumab including Imraldi (SB5), Cyltezo (BI 695501), Halimatoz (GP 2017), and Idacio (MSB11022) have been approved for patients with IBD, significantly expanding the treatment options for patients (Generics and Biosimilars Initiative, 2023a; Generics and Biosimilars Initiative, 2023b) (Table 2). In this part, we mainly discuss the most studied biosimilars of infliximab (Supplementary Table S1) and biosimilars of adalimumab in IBD (Supplementary Table S2).
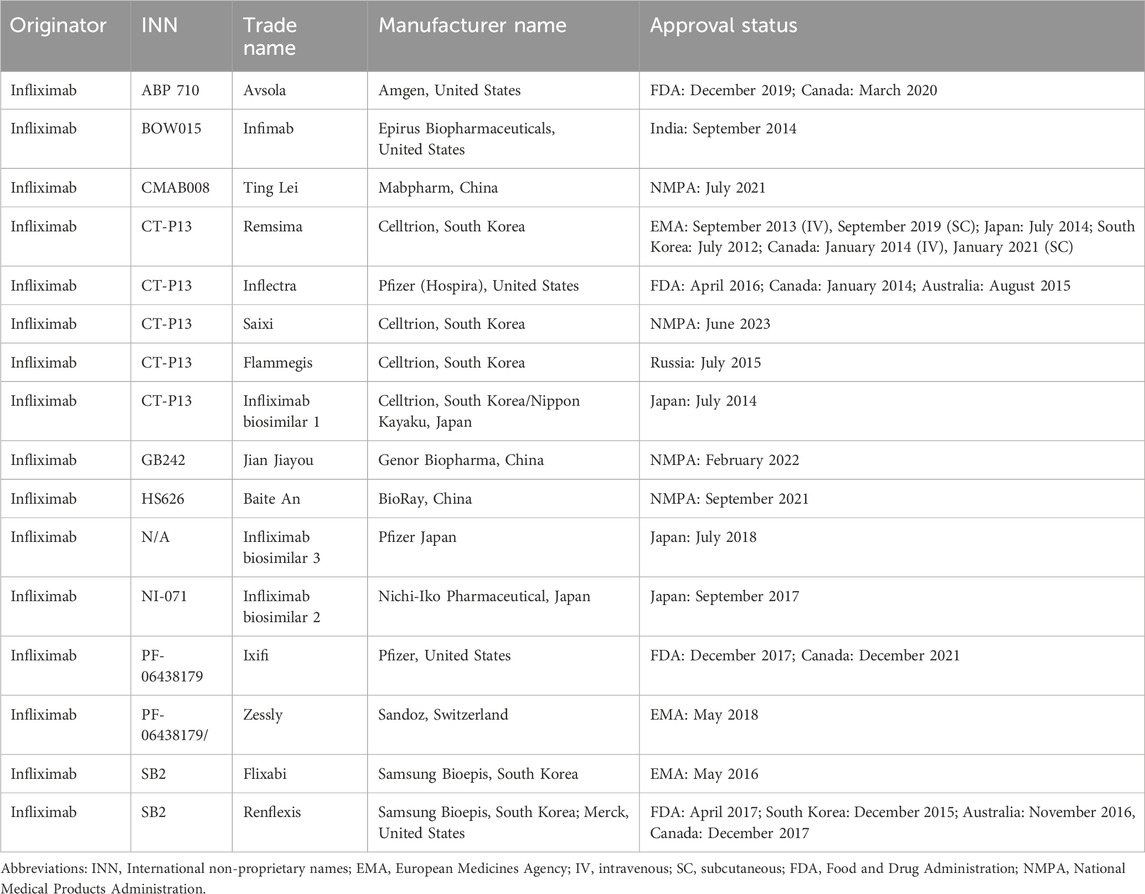
Table 1. Approval status of infliximab biosimilars.
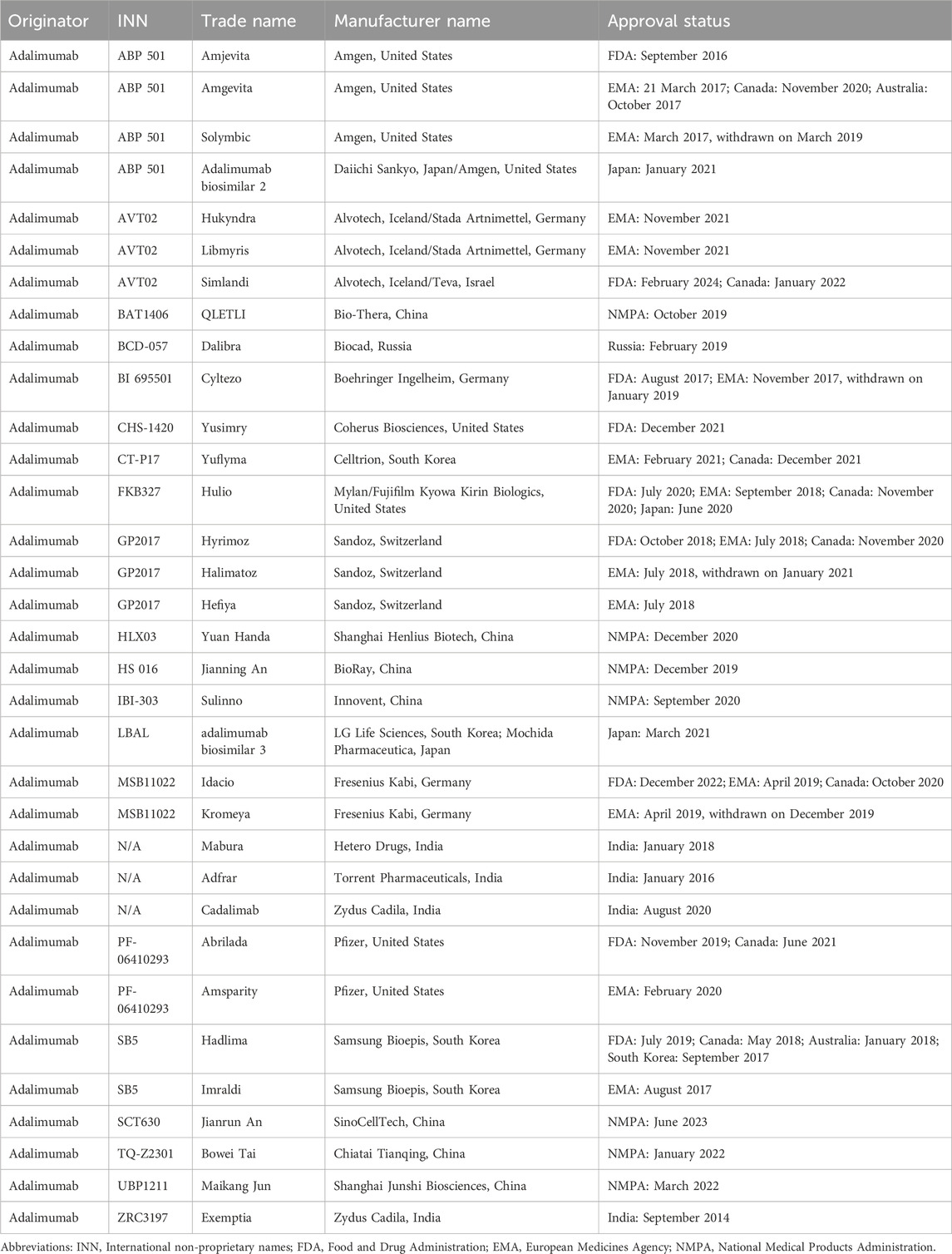
Table 2. Approval status of adalimumab biosimilars.
3.2 Biosimilars of infliximab in IBD3.2.1 CT-P13CT-P13 is the first approved biosimilar of infliximab used in immune-mediated diseases including RA, ankylosing spondylitis (AS), psoriasis, CD, and UC (Parigi et al., 2021). Two clinical head-to-head studies, the PLANETRA study, and PLANETAS study, demonstrated that CT-P13 (intravenous, IV formulation) was non-inferior to infliximab originator in terms of clinical efficacy and safety. Moreover, comparable pharmacokinetic and immunogenicity profiles have also been claimed in the two studies (Park et al., 2013; Yoo et al., 2013). Therefore, CT-P13 was approved for CD and UC based on extrapolation. The PROSIT-BIO cohort study of 547 patients with IBD firstly suggested that 73.7% of anti-TNF naïve patients with CT-P13 treatment could achieve clinical response at week 24, which was comparable with infliximab therapy (Fiorino et al., 2017). Following head-to-head comparison between infliximab and CT-P13 was conducted in 220 patients with active CD. The week 6 clinical response rates (a decrease of 70 points or more in CDAI, CDAI-70) were similar between the infliximab treatment group and the CT-P13 therapy group (74.3% vs. 69.4%). Furthermore, the two groups also showed comparable clinical remission rates and steroid-free remission rates at week 30. No significant differences in mean C reactive protein (CRP) concentrations, mean fecal calprotectin (FC) levels, pharmacokinetic, and pharmacodynamic profiles (Cmax and Ctrough) were observed between the two groups at every visit (Ye et al., 2019). Another comparative equivalence cohort study of 5050 infliximab naïve CD patients further proved the therapeutic equivalence between CT-P13 and infliximab (Meyer et al., 2019). Recently, a subcutaneous (SC) formulation of CT-P13 (CT-P13 SC) was developed for immune-mediated diseases. Although CT-P13 SC has its inherent shortcomings in comparison with the IV formulation of CT-P13 (CT-P13 IV), such as slower absorption, inadequate bioavailability, and lower initial peak concentrations, it shows its superiority in convenience, easy access, and time-saving (Bittner et al., 2018). It was claimed to be non-inferior to CT-P13 IV in patients with RA and IBD (Reinisch et al., 2019; Schreiber et al., 2021). An open-label, randomized, phase 1 study of 53 active CD and 78 active UC evaluated the efficacy and safety profiles of CT-P13 SC and suggested that the clinical response rates (86.8% vs. 74.4%, at week 30), clinical remission rates (60.5% vs. 38.5%, at week 30), and mucosal healing rates (47.7% vs. 30.8%, at week 22) were not significantly different between the CT-P13 SC group and the CT-P13 IV group. Besides, no differences in safety (treatment-emergent adverse events, TEAEs, 57.6% vs. 49.2%) and pharmacokinetics (Ctrough 21.45 μg/mL vs. Ctrough 2.93 μg/mL, at week 22) were found between the two arms, despite CT-P13 SC group showed numerically higher Ctrough during week 6 to week 54 (Schreiber et al., 2021). CT-P13 SC indeed provides an additional alternative for IBD patients. It holds the promise of reducing medical visit-associated costs, optimizing medical resources, and reducing the burden on the healthcare systems. It is also an important step towards patient empowerment and medication self-management in IBD treatment.
What should be noted is that switching from originator infliximab to biosimilar CT-P13 was also claimed to be safe and tolerated (Schmitz et al., 2018; Ye et al., 2019; Haifer et al., 2021). At week 54, the clinical response rates and clinical remission rates were similar between the continued treatment group and the switching treatment group (Ye et al., 2019). These results were in accord with similar findings by the pivotal NOR-SWITCH study that switching from infliximab to CT-P13 IV was not inferior to continued treatment with infliximab in patients with immune-mediated diseases including CD, UC, RA, and others (Jørgensen et al., 2017). The two groups presented similar disease worsening rates during 54-week follow-up (26% vs. 30%). Moreover, they also claimed no notable differences in trough drug concentrations in the two groups (Jørgensen et al., 2017). Comparable serum drug concentrations between the maintenance and the switching treatment group were also demonstrated in the SECURE study (Strik et al., 2018). Switching from originator infliximab to CT-P13 SC is also safe and tolerated (Smith et al., 2022). Concerns regarding safety and immunogenicity arose when we made a non-medical switch from originators to biosimilars. The NOR-SWITCH extension study of 380 patients with immune-mediated disease revealed that treatment switching did not increase the incidence of anti-drug antibodies (ADAbs) and AEs during 78-week follow-up (Goll et al., 2019). Several studies also demonstrated no differences in safety and immunogenicity between the maintenance and the switching treatment group (Jørgensen et al., 2017; Strik et al., 2018; Meyer et al., 2019; Ye et al., 2019). However, a contrary result that CT-P13 was inferior to infliximab was showed in another study (Chaparro et al., 2019). Chaparro et al. (Chaparro et al., 2019) claimed that switching treatment increased the risk of disease relapse in patients with IBD. Cautions need to be made when we interpret these results. Various definitions of disease relapse, disease remission, clinical remission, and disease worse were set in different studies. What’s more, the time for switching treatment from originators to biosimilars was also different, bringing additional hurdles to explain these findings. Further studies should be taken to elucidate these uncertainties.
3.2.2 SB2SB2 is the second infliximab biosimilar approved for CD and UC. One phase I study and another phase III clinical trial demonstrated its equivalence of pharmacokinetics, efficacy, and safety with originator infliximab in healthy volunteers and patients with RA, paving the way to SB2 approval in RA and other immune-mediated diseases (Shin et al., 2015; Choe et al., 2017). As for IBD, a prospective observational study assessed its efficacy and safety in 276 patients with IBD (136 CD and 140 UC). 57.3% of infliximab naïve patients can achieve steroid-free remission after an 8-week SB2 treatment, which is similar to the effectiveness of infliximab and CT-P13 (Macaluso et al., 2021b). One aspect should be taken into consideration is that previous anti-TNF treatment may decrease the efficacy of SB2 in IBD. In comparison with anti-TNF-naïve cases, patients who were previously exposed to anti-TNF presented lower steroid-free remission rates (66.1% vs. 40.0%) (Macaluso et al., 2021b). Another real-life study of 85 patients with IBD further verified its efficacy and immunogenicity. No significant differences in clinical remission rates, FC levels, and corticosteroid-free rates have been found after switching from infliximab to SB2 treatment (at a mean time of 329 days). Switching treatment also did not increase the risk of developing ADAbs and SAEs during a mean 135-day follow-up (Massimi et al., 2021). The long-term effectiveness, safety, and immunogenicity were further investigated by a German research. During an 80-week follow-up, the changes in the Harvey-Bradshaw Index (HBI) and partial Mayo Score (PMS) were not significant after switching treatment. Furthermore, about 72% of patients persisted in SB2 therapy at week 78, indicating that this switch was well tolerated (Fischer et al., 2021). The safety profile of SB2 in IBD varies between different studies. Some studies did not record any SAEs, while some other studies claimed that about 7.6%–20.7% of patients might suffer from SAEs (Fiorino et al., 2017; Fischer et al., 2021; Massimi et al., 2021; Bouhnik et al., 2023). The inconsistency in follow-up time might partly explain the difference in SAEs.
A single switch from infliximab to SB2 is claimed to be safe and tolerated in patients with IBD. Multiple switches from originators to CT-P13 to SB2 are still demonstrated to be safe and effective. An observational study evaluated the effects and pharmacokinetics of the first switch (from CT-P13 to SB2) and the second switch (from infliximab to CT-P13 to SB2) in 186 patients with IBD. No significant changes in CRP, HBI, or Simple Clinical Colitis Activity Index were found upon the first and second switches. Similar median Ctrough was recorded in pre-switch, early, and 1-year post-switch (4.9 μg/mL vs. 5.5 μg/mL vs. 5.3 μg/mL). Moreover, switching treatment did not exert a negative influence on disease response, given the comparable response rates during the 1-year follow-up (91% vs. 92% vs. 95%) (Luber et al., 2021). Another prospective multicenter cohort study of 176 patients with IBD further provided convincing evidence of efficacy and safety for multiple switches from originators to different biosimilars. The first switch (from CT-P13 to SB2) and the second switch (from infliximab to CT-P13 to SB2) showed comparable clinical remission rates at 12 months after switching treatment. Besides, 62.5% of the first switch group and 72.2% of the second switch group presented low FC levels (<250 mg/kg). It is worth noting that only the first switch group reported infusion reactions (3/80, 3.8%), suggesting multiple switches did not increase the risk of AEs (Hanzel et al., 2022). As aforementioned, the risk of increased immunogenicity is one of the core concerns when we make multiple switches. Available data demonstrated that no new ADAbs were developed after multiple switches (Hanzel et al., 2022). Although SB2 was claimed to be safe and effective in several studies, the clinical equivalence of SB2 in IBD is mostly proven in real-world studies, indicating a pressing need to conduct randomized, head-to-head, parallel clinical trials. Furthermore, few studies evaluated the efficacy of SB2 in achieving higher therapeutic goals, such as endoscopic mucosal healing and histologic remission. More studies are warranted to fill this gap.
3.2.3 PF-06438179/GP1111PF-06438179/GP1111 is another biosimilar of infliximab, which was approved for immune-mediated diseases by the FDA in 2017 and by the EMA in 2018 (Generics and Biosimilars Initiative, 2023a; Generics and Biosimilars Initiative, 2023b). One phase I clinical study evaluated the pharmacokinetics and immunogenicity of PF-06438179/GP1111 in 151 healthy subjects. The PF-06438179/GP1111 group showed great similarities in serum concentration-time profiles and ADAb response rates to the infliximab group (Palaparthy et al., 2018). The equivalent safety and efficacy of PF-06438179/GP1111 was demonstrated in a large randomized controlled trial of 650 patients with moderate to severe active RA (Cohen et al., 2018b). There are no significant differences in the American College of Rheumatology (ACR)-20, ACR-50, and ACR-70 response rates between the PF-06438179/GP1111 treatment group and the infliximab treatment group. Comparable Disease Activity Score (DAS) remission rates, and the 2010 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) remission rates were also claimed in this study. Besides, the PF-06438179/GP1111 arm showed similar all-cause TEAEs, incidence of ADAbs, and median Ctrough concentrations to the infliximab arm. When we made a non-medical switch from originator infliximab to PF-06438179/GP1111, efficacy was also well sustained in terms of ACR20, ACR50 and ACR70 response rates (Cohen et al., 2018b). This result indicated that single switch from a originator to PF-06438179/GP1111 was acceptable. Therefore, the strong equivalence of PF-06438179/GP1111 to infliximab with regard to efficacy, pharmacokinetics, and safety allowed the approval of it in the treatment of IBD, which is based on the concept of extrapolation. However, data on the efficacy and safety profiles in patients with IBD are very limited. A retrospective real-life study of 87 pediatric IBD patients assessed the efficacy of several biosimilars including CT-P13, SB2 and PF-06438179/GP1111, and demonstrated their favorable effectiveness in induction and maintenance of disease remission. Another single-center observational study reported that switching from SB2 to PF-06438179/GP1111 and re-switching from PF-06438179/GP1111 to SB2 were effective and tolerated (Macaluso et al., 2023). One point should be noted is that this study only included ten patients with IBD and followed up 16–28 weeks. The small sample size and short length of follow-up may limit the strength of conclusions. Further large, multicenter, long-term, prospective studies are needed. Moreover, head-to-head parallel studies are also warranted to provide more clinical evidence and clarify the exact role in the treatment of IBD, thus building confidence in the use of PF-06438179/GP1111 in IBD.
3.2.4 OthersABP 710, a biosimilar of infliximab, was approved for CD, UC, RA, AS, psoriasis, and psoriasis arthritis by the FDA in 2019 (Generics and Biosimilars Initiative, 2023b). It presented physicochemical, pharmacodynamic, and pharmacokinetic similarities to originator infliximab based on the analytical study and phase I clinical study (Chow et al., 2020; Saleem et al., 2020). Comparable efficacy, safety, and immunogenicity profiles were also demonstrated in the comparative clinical trial of RA. The ABP 710 group showed similar ACR-20 response rates (at week 22) to the infliximab arm (68.1% vs. 59.1%). Besides, there are also no clinically meaningful differences between the two arms in AEs (51.8% vs. 49.6%) and incidence of ADAbs (57.1% vs. 60.0%) (Genovese et al., 2020). No new safety signals have been reported. Other agents including NI-071, BOW015, GB242, CMAB008, etc. have also been approved by different countries. One network meta-analysis including seven randomized controlled trials of RA demonstrated that treatment of NI-071 was more probable to gain therapeutic success (the ACR-20 response rate), compared with ABP 710, CT-P13, PF-06438179/GP1111, and SB2 (Lee and Song, 2023). BOW015, GB242, and CMAB008 were claimed to be comparable to infliximab in terms of bioavailability, safety, and immunogenicity in three phase I clinical studies (Lambert et al., 2016; An et al., 2019; Zhang et al., 2019). Non-inferiority studies of these agents were all conducted in patients with RA. The ACR-20 response rates of the GB242 group and CMAB008 at week 30 were highly similar to that of the infliximab group (62.54% vs. 56.89%, and 57.6% vs. 62.2%, respectively). No clinically meaningful differences in safety, immunogenicity, and pharmacokinetics were found (Liu et al., 2022; Ye et al., 2023). However, studies on the above agents in IBD are still in the preliminary stages, no randomized studies and real-world data were reported. Further efforts should be made to facilitate clinical equivalence study in IBD.
3.3 Biosimilars of adalimumab in IBD3.3.1 ABP 501ABP 501, the first biosimilar of adalimumab, was approved for various diseases including RA, CD, UC, AS, and others by the FDA in 2016 and by the EMA in 2017(Generics and Biosimilars Initiative, 2023a; Generics and Biosimilars Initiative, 2023b). The analytical and functional characterization studies suggested that ABP 501 and adalimumab had great similarity in identity, general properties, physicochemical properties, purity and impurities, and inhibition effect on TNFα activities. The equivalent pharmacokinetics, similar safety profiles, and comparable immunogenicity of ABP 501 and adalimumab were further confirmed in a phase I study (Kaur et al., 2017). Based on the similar structures, functions, and pharmacokinetics between ABP 501 and adalimumab, further clinical equivalence studies were conducted. Comparable efficacy, safety, and immunogenicity between ABP501 and adalimumab were first demonstrated in patients with moderate to severe plaque psoriasis and then confirmed in cases with moderate to severe RA (Cohen et al., 2017; Papp et al., 2017). Following studies in IBD further claimed its favorable efficacy and safety. An observational study demonstrated that about 56% of CD patients could gain clinical remission upon ABP 501 treatment, with no new safety signals detected. Besides, the mean HBI scores (4.7 vs. 6.1) and CRP values (6.2 mg/L vs. 14.9 mg/L) at week 12 were numerically lower compared with the baseline values (Ribaldone et al., 2020). A three-arm propensity score-weighted analysis further compared the therapeutic effects and safety profiles of adalimumab and its biosimilars (ABP501 and SB5) in 86 CD and 69 UC patients. The three arms showed no significant differences in steroid-free clinical remission rates at induction stages (40.0% vs. 50.0% vs. 58.7%, at week 8) and maintenance stages (49.1% vs. 54.5% vs. 59.0%, at week 32). What should be noted is that superior efficacy was achieved by patients with CD compared with those with UC. The clinical response rate at week 8, and steroid-free clinical remission rates at weeks 8 and 32 were significantly higher in CD than in UC (Barberio et al., 2021). This is in accordance with the findings that adalimumab, infliximab, and its biosimilar were more effective in CD than UC, without no differences in safety and tolerability (Barberio et al., 2020). Underlying mechanisms are needed to be revealed.
ABP 501 seems to be as effective and tolerated as adalimumab in patients with IBD, thus providing an additional option for IBD patients who are naïve to or previously exposed to adalimumab. Switching from adalimumab to ABP 501 might be a cost-effective therapy for those patients. Available data indicated that there were no significant changes in HBI scores and CRP levels after switching from adalimumab to ABP 501 (Ribaldone et al., 2020). Similarly, Cingolani et al. (Cingolani et al., 2021) enrolled 55 IBD patients with switching treatment (adalimumab to ABP 501) and followed up for 6 months. In comparison with sustained therapy (adalimumab), switching treatment did not exert negative effects on HBI scores, PMS scores, and FC levels. There were still 76.3% of patients in remission after switching (Cingolani et al., 2021). Recently, the ADA-SWICTH study provided complementary data on disease relapse and safety profiles after switching treatment in patients with IBD (Casanova et al., 2023). Comparable relapse rates at 6 months (3% vs. 3%), 12 months (6% vs. 6%), and 24 months (26% vs. 12%) between switch treatment and sustained treatment group were recorded. The switching treatment group presented a numerally lower risk of suffering from endoscopic and/or radiologic activity compared with the other group (3% vs. 10%). Besides, this study also reported similar AEs between the two arms (6% vs. 5%), which further increased the confidence of physicians in the use of adalimumab biosimilars in clinical practice (Casanova et al., 2023). More valuable data were provided by the SPOSAB study. In this study, 85.5% of patients (adalimumab naïve) could gain clinical remission and 75.3% of patients could achieve a steroid-free remission after a 12-week ABP501 treatment. No efficacy difference was found between anti-TNFs-naïve patients and those previously exposed to anti-TNFs. However, inconsistent findings were reported by Cingolani et al. (2022). Better therapeutic effects of ABP 501 were achieved in anti-TNF-naïve patients, compared with anti-TNF-experienced ones (Cingolani et al., 2022). Different identifications of therapeutic effects in different studies may explain this inconsistency. One note in particular is that the incidence rates of SAEs were significantly lower in the switching therapy group. Thus, the lower incidence rates of SAEs might partly account for the finding that patients receiving switching therapy (adalimumab to ABP501) were more likely to persist in ABP 501 treatment, in comparison with those adalimumab-naïve patients (Macaluso et al., 2021a). Besides, no negative impacts of ABP 501 treatment on health-related quality of life were recorded, whether for the ABP 501 initiators or the adalimumab-ABP 501 switchers. More than 98% of physicians and patients expressed their satisfaction on ABP 501 treatment (Jin et al., 2024). Indeed, ABP 501 is truly effective and well-tolerated in IBD. However, data on immunogenicity, long-term efficacy, and long-term safety of ABP 501 in IBD are limited, suggesting a need to fill this gap. Cost-benefit analyses based on medical insurance of different countries are also warranted. Furthermore, in the “precision medicine” era, identifying suitable patients who will benefit most from ABP 501 is an essential prerequisite in the precision treatment of IBD. Therefore, exploring reliable biomarkers for predicting therapeutic response to ABP 501 is also needed.
3.3.2 SB5SB5 is another biosimilar of adalimumab, approved by the EMA in 2017 and the FDA in 2019(Generics and Biosimilars Initiative, 2023a; Generics and Biosimilars Initiative, 2023b). The clinical equivalence study was firstly conducted in a large phase III randomized study of 542 patients with moderate to severe RA. The ACR20, ACR50, and ACR70 response rates were equivalent between the SB5 treatment group and the adalimumab treatment group. No significant differences in the incidence of TEAEs, development of ADAbs, and pharmacokinetics were reported in this study (Weinblatt et al., 2018). By extrapolation, the approval was extended to IBD, axial spondylarthritis, and psoriasis arthritis (Müller-Ladner et al., 2023).
Lukas et al. (2020) firstly provided the real-life study that directly compared the efficacy, safety, pharmacokinetic, and immunogenicity profiles between the originator and SB5. 93 IBD patients received switch treatment (from adalimumab to SB5) and the other 93 patients still received originator adalimumab therapy. The two groups did not show any significant changes at week 10 with regard to HBI scores, PMS scores, CRP levels, and FC concentrations. They also claimed no notable differences in trough drug concentrations (13.0 μg/mL vs. 13.7 μg/mL) and the incidence of ADAbs (2% vs. 2%) between the two arms at week 10. However, the follow-up time was only 10 weeks, too short to evaluate the long-term safety profiles. Further study conducted by Barberio et al. (2021) provided additional information on the long-term efficacy and safety profiles in patients with IBD. They compared the effectiveness and safety profiles of SB5 and adalimumab at weeks 8 and 48. The rates of steroid-free clinical remission at the two time points were 58.7% and 59.0%, which were comparable to the rates of adalimumab (40% and 49.1%, respectively). Similar clinical response rates at weeks 8 and 48 were also collected in this study (Barberio et al., 2021). Data on the 1-year performance of SB5 in patients with IBD were further reported by a UK study. They divided patients into two arms, the SB5-switch group and the SB5-start group (adalimumab naïve), and followed up at a median time of 13.7 months and 8.3 months, respectively. SB5 showed comparable effectiveness to adalimumab given similar 1-year drug persistence rates (62.5% vs. 50.89%) (Chen et al., 2019; Derikx et al., 2021). Switching treatment also did not worsen the biochemical remission rates, fecal biomarker remission rates, and clinical remission rates at weeks 26 and 52. Besides, there were also no differences in the median Ctrough concentrations at weeks 0, 26, and 52 after switching treatment (10.1 μg/mL vs. 11.6 μg/mL vs. 7.8 μg/mL). This is consistent with the other two studies reporting stable trough drug concentrations after switching from adalimumab to SB5 (Lukas et al., 2020; Tapete et al., 2022).
About 19.9% of patients in the SB5-switch cohort and 17.3% of patients in the SB5-start cohort reported AEs, respectively. The most common AE in the SB5-switch cohort was injection-site pain (66.7%), which lead to a double-switch treatment (from adalimumab to SB5 to ABP 501) in these patients. Therefore, this study provided the first data on the double-switch treatment. Median trough concentrations were stable during the first and the second switch treatment, suggesting that multiple switches might work in cases intolerant to SB5 (Derikx et al., 2021). Similarly, switching from adalimumab to ABP 501 to SB5 was also tolerated (Ribaldone et al., 2021). It did not impair the efficacy and increase the risk of AEs in patients with IBD. Given that injection-site pain negatively affected treatment persistence, solutions to help relieve injection-site pain were designed. A citrate-free and high concentration of SB5 (SB5-HC) was claimed to be associated with less injection site pain (Ahn et al., 2022). Besides, injection technique training and psychological interventions are also important to alleviate pain. Overall, SB5 is effective and safe in IBD, though this conclusion was based on post-marketing evidence. Healthcare professionals and patients expressed their concerns about its efficacy and safety, causing some negative effects on the market share of SB5. Therefore, further randomized controlled clinical trials may help build confidence and increase the uptake of SB5. Moreover, some studies did not perform dose optimization in a standardized manner and evaluate endoscopic/histological healing after dose optimization, which might cause potential selection bias.
3.3.3 BI 695501BI 695501 is another biosimilar of adalimumab (Wynne et al., 2016). The regulatory approval of BI 695501 was granted in Europe and the United States in 2017 based on the “totality of the evidence” (Generics and Biosimilars Initiative, 2023a; Generics and Biosimilars Initiative, 2023b). The bioequivalence, comparable safety, and similar immunogenicity of BI 695501 to adalimumab were first demonstrated in a phase I study of 327 healthy volunteers in 2016 (Wynne et al., 2016). Two years later, the efficacy data were primarily obtained in patients with RA (Cohen et al., 2018a). This study suggested the non-inferiority of BI 695501 to originator adalimumab in terms of efficacy, safety, and immunogenicity. Switching from adalimumab to BI 695501 was not associated with lower efficacy, increased incidence of AEs, and elevated levels of ADAbs (Cohen et al., 2018a). By extrapolation, the approval was extended to other indications including CD, UC, AS, psoriasis, and others.
Clinical data on BI 695501 in patients with IBD were limited. One large, multicenter, randomized, double-blind, phase 3 study including 147 moderately to severely active CD patients divided patients into two groups, the BI 695501 group and the adalimumab group (Hanauer et al., 2021). The two groups showed similarities in clinical response rates (90% vs. 94% at week 4, and 81% vs. 82% at week 24), clinical remission rates (68% vs. 75% at week 24), and AE rates (63% vs. 56% at week 24). Besides, this VOLTAIRE-CD study also evaluated the feasibility of switching from adalimumab to BI 695501. No negative impacts of switching treatment on efficacy and AEs were claimed. Patients in the switch group presented a similar degree of reduction in CDAI scores and a similar incidence of TEAEs to those in the BI 695501 sustained group (Hanauer et al., 2021). Likewise, the VOLTAIRE-X study of 238 patients with chronic plaque psoriasis further demonstrated that switching back and forth from adalimumab to BI 695501 was safe, effective, and tolerated (Menter et al., 2022). This study provided direct evidence for the interchangeability of BI 695501. Thus, BI 695501 (Cyltezo) became the first FDA-approved interchangeable biosimilar to adalimumab (Kay et al., 2024). This indicated that pharmacists can substitute the biosimilar for its originator without the permission of the prescribing healthcare professionals (Alvarez et al., 2020). The “interchangeable” logo may greatly increase the uptake of Cyltezo. More treatment options are thus provided for patients who need repeated therapy during the overall disease course. However, the paucity of safety and immunogenicity data on IBD highlighted the need to conduct real-world studies in the future. Besides, evaluating the long-term outcomes of BI 695501 on the basis of interchangeability designation in different diseases is also warranted.
3.3.4 GP2017GP2017 is the fourth adalimumab biosimilar approved by the EMA and the third one approved by the FDA in 2018 (Generics and Biosimilars Initiative, 2023a; Generics and Biosimilars Initiative, 2023b). The equivalent efficacy, safety, and immunogenicity between GP2017 and adalimumab were demonstrated in a phase III randomized study of 465 patients with plaque psoriasis. This study also demonstrated that multiple switches between adalimumab and GP2017 did not impair the disease outcomes and affect the safety and immunogenicity profiles (Blauvelt et al., 2018). The following study in patients with moderate to severe active RA further confirmed the non-inferiority of GP2017 to adalimumab in terms of efficacy, safety, and immunogenicity (Wiland et al., 2020). GP2017 was approved for IBD through extrapolation of indications.
Real-life data on the efficacy of GP2017 in IBD were provided by an Italy study (Mocci et al., 2022). This study retrospectively analyzed the clinical data of 134 patients with IBD. Among these patients, 62 patients received GP2017 treatment while the others received adalimumab therapy. Similar clinical remission rates and clinical response rates were reported regardless of whether they were naïve to biologics or not. 82.3% of patients in the GP2017 group and 75.0% of patients in the adalimumab group achieved clinical remission at a median follow-up time of 12 months. No clinically meaningful differences in the rates of treatment optimization and surgery, as well as the incidence of AEs were suggested. More importantly, GP2017 showed better effects in achieving mucosal healing than adalimumab. The mucosal healing rate in the GP2017 group was about 1.5 times as that in the adalimumab group (89.2% vs. 60.2%). Recently, another real-world retrospective study evaluated the impacts of switching treatment in IBD patients (Vernero et al., 2023). Switching from adalimumab to GP2017 did not increase the clinical disease activity and interfere the treatment persistence. Patients who were previously exposed to infliximab were at a higher risk of needing dose optimization of GP 2017 (Vernero et al., 2023). However, the retrospective design cannot prove the causal association and control the potential confounding factors. Well-designed, well-paired, prospective studies might add useful information. A prospective observational study of 50 IBD patients further proved the favorable efficacy and safety profiles of GP2017. 75.0% of patients obtained remission or partial response after 12-week treatment of GP2017. A median decrease of CDAI and Mayo score was 140.5 and 4.0. respectively (Wasserbauer et al., 2022). This study also had limitations, including a lack of reference product control, a short follow-up time, and a small sample size. Recently, a cross-sectional, questionnaire-based study assessed the subjective efficacy of switching treatment in 179 IBD patients (Sarlós et al., 2023). Patients with GP2017 switching treatment reported better efficacy of GP2017 than adalimumab. However, they also complained of a higher incidence of new AEs (1.79 per patient) that did not occur during adalimumab treatment. Most of these patients also expressed their willingness to switch back to adalimumab if possible (Sarlós et al., 2023). Such a contradiction may be partly explained by the “nocebo” effect, an unfavorable therapeutic effect of a medical therapy that is not caused by pharmacological effects and is related to patients’ high expectations on it (Colloca et al., 2019).
Overall, GP2017 is as effective and safe as adalimumab in patients with IBD. However, there is relatively limited data on the pharmacokinetics and immunogenicity of GP2017 in IBD. Little is known about the drug concentrations and ADAb levels after switching treatment. More prospective studies are also needed to evaluate the performance of multiple switches between adalimumab and GP2017 in patients with IBD.
3.3.5 OthersBiosimilars of adalimumab including FKB327, MSB11022, AVT02, PF-06410293, CHS-1420, CT-P17, and others were also approved for treatment of CD and UC (Generics and Biosimilars Initiative, 2023b; Generics and Biosimilars Initiative, 2023a). However, most clinical evidence was obtained from patients with RA and plaque psoriasis. There were relatively limited efficacy and safety data on them in IBD. FKB327 treatment showed high efficacy in inducing and maintaining disease remission or partial response at week 12 (18/22, 81.8%), which was comparable to the effectiveness of GP2017 (21/27, 75.0%) (Wasserbauer et al., 2022). A large, multicenter, observational study of 533 IBD patients evaluated the efficacy and safety profiles of four biosimilars of adalimumab (SB5, APB501, GP2017, and MSB11022). Available data indicated that 81.8% of patients with MSB11022 could achieve clinical remission, similar to SB5 (75.2%), APB501 (78.3%), and GP2017 (77.5%). MSB11022 also showed similarities in steroid-free remission rates and mucosal healing rates to the other three biosimilars. No new safety concerns were identified in MSB11022 (Tursi et al., 2023). However, the data must be viewed critically because the patients included in the MSB11022 group were only 11, which may weaken the strength of the evidence. Another Italy study of 143 IBD patients further compared the efficacy and safety of the fo
留言 (0)